
Autor: dr Adam Jarmuła, Instytut Biologii Doświadczalnej im. M. Nenckiego PAN
Dokowanie molekularne jest metodą pozwalającą badać rozpoznanie molekularne jednej cząsteczki przez inną, a więc przewidywać sposoby wiązania oraz powinowactwo do wiązania w kompleksie utworzonym przez dwie lub więcej cząsteczek o znanych strukturach. Dokowanie w układzie białko-ligand jest ważnym rodzajem dokowania molekularnego znajdującym zastosowanie w nowoczesnym projektowaniu leków w oparciu o strukturę. Może być ono rozumiane jako efektywna i ekonomiczna alternatywa dla eksperymentalnych metod określania/prognozowania sposobów wiązania i powinowactwa do wiązania w kompleksie pomiędzy białkowym receptorem i ligandem.
Podstawy teoretyczne
W przypadku receptora białkowego (RB) i ligandu (L) , celem dokowania jest poprawne przewidzenie sposobu wiązania w kompleksie RBL w warunkach równowagi w roztworze wodnym opisanej równaniem (1)
(1) [RBL]↔ [RB] + [L]
Miarą stabilności powstającego kompleksu RBL jest energia swobodna wiązania ΔGbind, która związana jest z powinowactwem do wiązania albo inaczej stałą asocjacji kompleksu wedle formuły: ΔGbind = -RT ln(Ka), gdzie R to stała gazowa, T – temperatura, zaś Ka – stała asocjacji równa ilorazowi stężenia kompleksu RBL do stężeń wolnych składników RB oraz L (Ka = [RBL]/[RB][L]). Predykcja aktywności biologicznej wymaga informacji na temat sposobu wiązania w kompleksie RBL, którą uzyskuje się porównując wartości ΔGbind dla różnych sposobów wiązania (póz) ligandu L.
Procedura dokowania
Dokowanie liganda do receptora białkowego obejmuje dwa następujące po sobie procesy: próbkowanie (sampling) i oceny punktowe (scoring). Próbkowanie polega na generowaniu różnych konformacji ligandu w miejscu wiążącym receptora. Uzyskuje się to uwzględniając zarówno próbkowanie konformacyjne ligandu oraz elastyczność (ograniczoną zmienność) receptora białkowego. Oceny punktowe odnoszą się do przewidywania powinowactwa do wiązania indywidualnych konformacji ligandu. W ten sposób ustalana jest tzw. lista rankingowa, klasyfikująca cząsteczki ligandów w kolejności od największego do najmniejszego prognozowanego powinowactwa. Najlepiej oceniania poza (charakteryzująca się najkorzystniejszą prognozowaną energią swobodną wiązania ligandu do receptora lub jej przybliżeniami stosowanymi w funkcjach oceniających) jest uznawana za preferowany sposób wiązania w kompleksie receptor-ligand.
Metody dokowania
Metody opierające się na próbkowaniu konformacyjnym ligandu obejmują: dopasowanie kształtów ligandu i miejsca wiążącego w receptorze białkowym, poszukiwanie systematyczne oraz algorytmy stochastyczne. Z kolei próbkowanie konformacyjne białka bywa uwzględniane poprzez: miękkie dokowanie, relaksację molekularną, dokowanie z uwzględnieniem elastyczności łańcuchów bocznych oraz z udziałem wielu konformacji receptora. Stosowane funkcje oceniające mają charakter empiryczny lub oparte są na wiedzy bądź wyprowadzone z wykorzystaniem pól siłowych mechaniki molekularnej. Poniżej przedstawię pokrótce poszczególne metody.
Próbkowanie ligandu
Najprostsza metoda samplingu ligandów polega na dopasowaniu powierzchni molekularnych dokowanego ligandu i miejsca wiążącego w białku. Oczywistymi zaletami tej metody są prostota oraz wydajność obliczeniowa, wadą – sztywna konformacja ligandu podczas dopasowywania jego kształtu do kształtu receptora (zazwyczaj, by przeciwdziałać temu ograniczeniu, proces jest przeprowadzany z użyciem wielu pre-generowanych konformacji ligandu). Kolejną metodą jest poszukiwanie systematyczne, które zakłada wygenerowanie wszystkich możliwych konformacji wiążących ligandu poprzez eksplorację jego wszystkich stopni swobody. W ramach metody stosuje się próbkowanie w oparciu o systematyczną rotację (o zadaną wartość) wszystkich wiązań ligandu zdolnych do rotacji. Pomimo swej kompletności, taki algorytm staje się niepraktyczny ze wzrostem liczby wiązań zdolnych do rotacji z uwagi na zbyt dużą ilość kombinacji do policzenia. Praktyczną alternatywą jest przeprowadzenie początkowego poszukiwania z więzami nałożonymi na niektóre wiązania ligandu zdolne do rotacji, a następnie dodatkowego poszukiwania z użyciem protokołu optymalizacyjnego. Innym podejściem wykorzystywanym w metodzie systematycznego poszukiwania jest tzw. fragmentacja, w ramach której ligand dzielony jest na szereg sztywnych fragmentów, dokowanych w różnych orientacjach do miejsca wiążącego w receptorze, i tam łączonych kowalencyjnie, co prowadzi do powstania unikalnych póz ligandu. Jeszcze innym algorytmem jest próbkowanie przy udziale wielu pre-generowanych, sztywnych konformacji ligandu wystepujących w różnych orientacjach (tzw. conformational ensemble). Inną grupą metod samplingu ligandów są algorytmy stochastyczne, w których próbkowanie polega na przypadkowych perturbacjach w konformacyjnej, rotacyjnej i translacyjnej przestrzeni ligandu. Zmiany są akceptowane lub odrzucane w zależności od kryterium probabilistycznego. Pośród algorytmów stochastycznych można wymienić metodę Monte Carlo, metody przeszukiwania z Tabu, optymalizację wielocząsteczkową (rojem cząsteczek) oraz algorytmy ewolucyjne.
Próbkowanie receptora białkowego
Związanie ligandów powoduje zmiany konformacyjne w białku (tzw. indukowane dopasowanie), które mogą mieć bardzo różny zakres: od niewielkich zmian w łańcuchach bocznych do masywnych przemieszczeń dużych domen. Elastyczność białka jest trudna do uwzględnienia w procesie dokowania z uwagi na często duże rozmiary cząsteczek i związaną z tym olbrzymią ilość stopni swobody receptora białkowego.
Podstawową metodą próbkowania przestrzeni konformacyjnej białka jest tzw. miękkie dokowanie, dopuszczające małe nałożenia pomiędzy powierzchniami van der Waalsa atomów białka i ligandu. Zaletą tej metody jest zapobieganie konfliktom sterycznym przy równoczesnym zachowaniu określonych póz ligandu, które mogą okazać się poprawne. Metoda jest wydajna obliczeniowo i wygodna w implementacji. Z drugiej strony, miękkie dokowanie może zwiększać ilość niepoprawnych póz, które zostały wstępnie zaakceptowane. Prócz tego, jest niewystarczające w przypadku większych zmian konformacyjnych. Alternatywną metodą jest próbkowanie konformacyjne łańcuchów bocznych, podczas którego łańcuchy boczne ulegają rotacjom, gdy szkielet białka pozostaje nieruchomy. Wczesne próby uwzględnienia próbkowania łańcuchów bocznych były skupione na wykorzystywaniu bibliotek rotamerów łańcuchów bocznych aminokwasów oraz algorytmów znajdujących kombinacje konformerów łańcuchów bocznych i orientacji ligandu obdarzone najniższą energią. Nowsze podejścia inkorporują ciągłą lub dyskretną elastyczność łańcuchów bocznych. Zalicza się tutaj podejście wykorzystujące algorytm genetyczny, które uwzględnia obserwowane eksperymentalnie konformacje białka oraz pełną elastyczność ligandu, podejście wykorzystujące tzw. symulowane wyżarzanie (simulated annealing), które uwzględnia przypadkowe zmiany konformacji łańcuchów bocznych akceptowane w oparciu o kryterium probabilistyczne (np. Metropolis w metodzie Monte Carlo) oraz podejście wykorzystujące hipotezę najmniejszej rotacji, gdzie małe rotacje kątów torsyjnych (w zakresie poniżej 30 stopni) są faworyzowane w stosunku do większych rotacji. Ogółem, próbkowanie konformacyjne łańcuchów bocznych modeluje dość dobrze umiarkowane zmiany konformacyjne, jest natomiast niedostateczne w przypadku dużych zmian konformacyjnych z powodu nieuwzględniania elastyczności fragmentów białka należących do szkieletu. Kolejną metodą próbkowania przestrzeni konformacyjnej białka, która częściowo eliminuje to niedomaganie, jest relaksacja molekularna. W tej metodzie ligand jest dokowany w miejscu wiążącym receptora w postaci sztywnej, a następnie atomy szkieletu i łańcuchów bocznych w miejscu wiążącym i jego bliskim otoczeniu są poddawane relaksacji celem ustalenia równowagi konformacyjnej. Kolizje steryczne pojawiające się na etapie sztywnego dokowania eliminowane są podczas relaksacji dzięki poszerzonemu próbkowaniu przestrzeni konformacyjnej przy użyciu symulacji dynamiki molekularnej lub Monte Carlo. Uwzględniając oprócz próbkowania łańcuchów bocznych także próbkowanie fragmentów szkieletu białka, metoda relaksacji molekularnej umożliwia modelowanie większych zmian konformacyjnych niż wcześniej omówione metody próbkowania przestrzeni konformacyjnej białek. Zarazem metoda ta jest bardziej wymagająca w doborze funkcji oceniających oraz bardziej kosztowna obliczeniowo. Ostatnią z głównych metod próbkowania przestrzeni konformacyjnej receptora białkowego jest dokowanie z udziałem wielu konformacji białka. W tej metodzie elastyczność białka jest modelowana z użyciem wielu struktur białka w różnych konformacjach. Mogą to być struktury krystalograficzne, pozyskane z badań NMR oraz z symulacji dynamiki molekularnej, Monte Carlo lub symulowanego wyżarzania. Poszczególne struktury mogą być ze sobą mieszane w ten sposób, że podobne części struktur są łączone w jedną średnią strukturę, podczas gdy części różniące się są traktowane jako alternatywy. Zaletą metody jest poszerzenie przestrzeni konformacyjnej dla molekularnego rozpoznania ligandu, mankamentem konieczność oddzielnego dokowania dla indywidualnych konformacji białka, wydłużająca proces. By rozwiązać ten problem, proponowane jest m.in. podejście, w którym poszczególne konformery białka są nakładane na siebie, a następnie ich siatki scalane we wspólny obiekt (tzw. 4D grid), który wykorzystuje się prowadząc pojedynczą symulację dokowania.
Funkcje oceniające
Wśród funkcji oceniających da się wyróżnić następujące rodzaje: empiryczne, które sumują ważone empiryczne człony energii (wagi członów są dostosowywane tak aby odtwarzać powinowactwa do wiązania związków ze zbioru treningowego), oparte na wiedzy, które opierają się na założeniu, że oddziaływania występujące często w kompleksach białko-ligand obecnych w bazach danych zwiększają wzajemne powinowactwo białka i ligandu (sprzyjają utworzeniu kompleksu) oraz wykorzystujące pola siłowe mechaniki molekularnej, które sumują udziały dwóch rodzajów energii – oddziaływania pomiędzy receptorem i ligandem oraz zmiany wewnętrznej energii ligandu (związanej z jego naprężeniem sterycznym indukowanym podczas wiązania przez receptor). Dodatkowy komentarz poświęcę tej ostatniej metodzie. Standardowe pola siłowe zostały oryginalnie sformułowane do opisu oddziaływań entalpowych w fazie gazowej, przeto nie uwzględniają one efektów solwatacyjnych oraz członów entropowych. Zazwyczaj obliczane są oddziaływania van der Waalsa (steryczne) reprezentowane przez potencjał Lennarda-Jonesa oraz elektrostatyczne opisane potencjałem Coulomba. Efekty desolwatacyjne są modelowane dodatkowo, przy użyciu jednej z następujących metod: prostego algorytmu, w którym efekty rozpuszczalnika są modelowane poprzez zmianę wartości stałej dielektrycznej w zależności od odległości pomiędzy oddziałującymi ładunkami (model przesadnie uproszczony, energie niedokładne), metod ”alchemicznych”, uwzględniających wprost (explicite) obecność cząsteczek wody (FEP, TI, bardzo dokładne energie, wysoki koszt obliczeniowy, ograniczona możliwość stosowania) lub metod ciągłych, uwzględniających obecność wody implicite, tj. traktujących wodę jako jednorodny ośrodek dielektryczny (PB/SA, GB/SA, umiarkowanie dokładne energie, umiarkowany koszt obliczeniowy). Obliczenia entropii dla dużych układów molekularnych pochłaniają bardzo dużo czasu, zaś analiza drgań normalnych (Normal Mode Analysis) która bywa stosowana w tym celu, umożliwia jedynie zgrubne oszacowania obwarowane znacznymi marginesami błędu.
Ostateczny wybór pozy wiążącej dokonywany jest często na zasadzie konsensusu pomiędzy różnymi funkcjami oceniającymi.
Oprogramowanie
Istnieje duży wybór programów do dokowania molekularnego, zarówno komercyjnych jak i swobodnie dostępnych. Najbardziej znane/cieszące się największą renomą programy to m.in. Autodock, Autodock Vina, DOCK, DockVision, GOLD, Glide, ICM, FlexX, FRED, PSI-DOCK oraz PSO@AutoDock. Bardziej lub mniej szczegółowe listy takiego oprogramowania można znaleźć w wielu miejscach w sieci, poniżej podaję adres jednej z takich stron, zawierającej zarówno wyczerpującą listę oprogramowania (wraz z krótkimi charakterystykami) oraz listę serwerów oferujących usługi w zakresie dokowania: http://www.click2drug.org/index.html#Docking
Dokowanie w wirtualnym przeszukiwaniu
Wirtualne przeszukiwanie (Virtual Screening) jest metodą komputerową przeszukiwania przestrzeni chemicznej używaną w racjonalnym projektowaniu leków. W ramach metody, przeprowadzana jest szybka komputerowa eksploracja molekularnych baz danych 3D (przestrzennych), której zadaniem jest pozyskiwanie nowych związków wiodących w systemie projektowania leków. Jedną z głównych technik znajdujących zastosowanie w wirtualnym przeszukiwaniu jest dokowanie molekularne, które pozwala prognozować powinowactwo do celu molekularnego związków chemicznych z molekularnych baz danych testowanych jako kandydaci na leki (Rysunek 1).
Małe cząsteczki zawarte w bazach molekularnych 2D (ligandy) są wstępnie filtrowane przy użyciu zdefiniowanych reguł (np. tzw. Reguła 5 Lipińskiego), następnie dodawane są atomy wodoru, przypisywane ładunki cząstkowe oraz cząsteczki są konwertowane do postaci 3D. W ten sposób przefiltrowane związki, występujące jako struktury trójwymiarowe, są dokowane z użyciem programów komputerowych do trójwymiarowej struktury białka będącego celem molekularnym (receptorem). Trójwymiarowa struktura receptora białkowego jest pozyskiwana zwykle z bazy danych Protein Data Bank. Efekty dokowania są oceniane przy pomocy funkcji oceniających i najlepiej ocenione ligandy, najkorzystniej oddziałujące z receptorem białkowym w utworzonym kompleksie, są typowane jako związki wiodące (leads). Na bazie związków wiodących projektowanych jest szereg analogów, ulepszonych inhibitorów celu molekularnego o założonej zwiększonej aktywności i/lub specyficzności względem celu. W procesie optymalizacji aktywności wykorzystywane jest budowanie modeli zależności pomiędzy strukturą i aktywnością (QSAR), które pozwalają na przewidywanie zmian w aktywności wskutek powiązanych zmian w strukturze. Pomaga to w racjonalnym projektowaniu zmian strukturalnych w cząsteczkach ligandów. Najbardziej obiecujące związki są kierowane do testów biologicznych; najlepsze z nich mogą być dopuszczone do badań klinicznych i w perspektywie zostać zaaprobowane jako lek. Procedura wirtualnego przeszukiwania może być stosowana wielokrotnie, również naprzemiennie z inną, tzw. procedurą wysokoprzepustowego przeszukiwania (High-Throughput Screening).
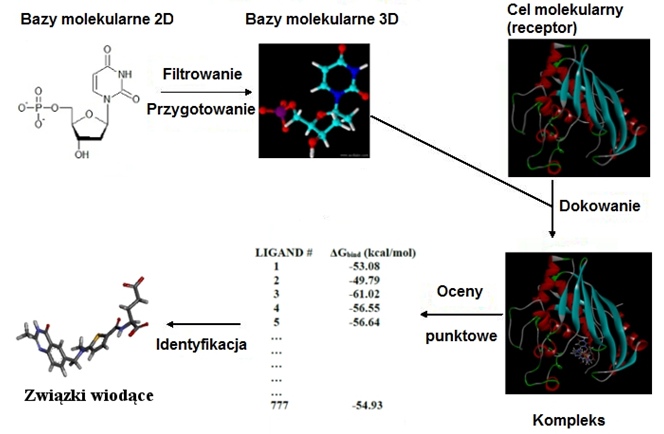
Rysunek 1. Schemat metody wirtualnego przeszukiwania.
Stan obecny i przyszłe wyzwania
Dokowanie molekularne w układzie receptor białkowy-ligand w połączeniu z innymi technikami modelowania komputerowego, a w szczególności dynamiką molekularną i wirtualnym przeszukiwaniem, jest zyskującą na znaczeniu metodą identyfikowania związków wiodących w projektowaniu leków. W ostatniej dekadzie metoda rozwija się bardzo intensywnie, systematycznie pojawia się nowe oprogramowanie, ulepszane są dotychczasowe algorytmy. Największy postęp w procesach dokowania został dokonany w zakresie próbkowania ligandu, podczas gdy próbkowanie białka pozostaje centralnym problemem dokowania z uwagi na ogromne ilości stopni swobody białka potrzebne do uwzględnienia i niewystarczające moce komputerowe aby to w pełni osiągnąć. Ocena zmian entropii towarzyszących dokowaniu pozostaje najważniejszym wyzwaniem w rozwoju funkcji oceniających. Wyzwaniem pozostaje również uwzględnienie w procesie dokowania ”strukturalnych” cząsteczek wody, uczestniczących w wiązaniach wodorowych z białkiem i/lub ligandem.
Literatura
1. Huang, S.-Y., Zou, X. Int. J. Mol. Sci. 2010, 11, 3016-3034.
2. Cheng, T., Li, Q., Zhou, Z., Wang, Y., Bryant, S.H. AAPS J. 2012, 14, 133-141.
3. Bello, M., Martinez-Archundia, M., Correa-Basurto, J. Expert Opin. Drug Discov. 2013, dx.doi.org/10.1517/17460441.2013.794780
4. Elokely, K.M., Doerksen, R.J. J. Chem. Inf. Model. 2013, dx.doi.org/10.1021/ci400040d
5. Sousa, S.F., Ribeiro, A.J., Coimbra, J.T., Neves, R.P., Martins, S.A., Moorthy, N.S., Fernandes, P.A., Ramos, M.J. Curr. Med. Chem. 2013, 20, 2296-2314.