PRZEDRUK, oryginał dostępny pod adresem www
Tytuł oryginalny: Materiały dodatkowe – kwasy i pochodne
Autor: dr Marek Żylewski
Uniwersytet Jagielloński (www)
Wydział Farmaceutyczny Collegium Medicum (www)
Katedra Chemii Organicznej (www)
Kierownik: Prof. UJ, dr hab. Marek Cegła
Adres:
ul. Medyczna 9
30-688 Kraków
Kontakt: tel. 012 620 55 00
Hydroliza estrów
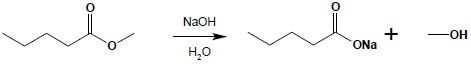
Hydrolizę estrów można wykonać zarówno w środowisku kwaśnym jak i zasadowym. Reakcja biegnąca w środowisku kwaśnym jest dokładnym odwróceniem reakcji bezpośredniej estryfikacji, jak to już zostało wspomniane i oczywiście jest procesem równowagowym. Hydroliza estrów przebiegająca w środowisku zasadowym (zmydlanie estrów) jest procesem nieodwracalnym, w którego wyniku powstaje sól kwasu i odpowiedni alkohol:
Transestryfikacja (alkoholiza estrów)
Reakcja estrów z alkoholami jest równowagowym procesem pozwalającym na otrzymanie nowego estru:
![]()
Aby osiągnąć dobre wydajności, stosuje się albo znaczny nadmiar jednego z substratów, albo oddestylowuje się lotny produkt reakcji.
Aminoliza estrów
Estry są cennymi środkami acylującymi aminy. Reakcje z reguły przebiegają z dobrymi wydajnościami, ale ze względu na mniejszą reaktywność estrów niż chlorków czy bezwodników wymagają dłuższego czasu prowadzenia procesu:

Reakcję tą stosuje się w praktyce, kiedy odpowiednie chlorki czy bezwodniki są trudne do uzyskania.
Reakcja estrów ze związkami Grignarda
Estry reagują ze związkami magnezoorganicznymi analogicznie do chlorków czy bezwodników. Reakcja rozpoczyna się od addycji cząsteczki związku Grignarda do grupy karbonylowej estru, a następnie ulega odłączeniu cząsteczka alkoholanu:
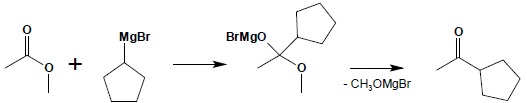
Powstały keton natychmiast reaguje z drugą cząsteczką związku Grignarda:
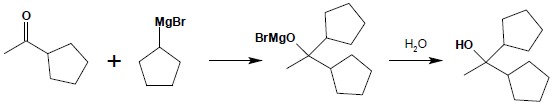
Redukcja estrów
Redukcja grupy estrowej zachodzi dość trudno. W jej wyniku otrzymuje się alkohol, będący wynikiem redukcji fragmentu pochodzącego od kwasu oraz uwolniony zostaje alkohol wchodzący w skład estru:
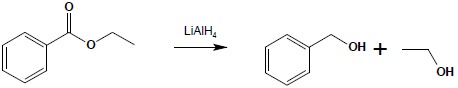
Do redukcji estrów stosować można LiAlH4 jak również dobre wyniki uzyskuje się redukując estry za pomocą sodu w alkoholu. Przed odkryciem LiAlH4 ta ostatnia metoda była główną reakcją stosowaną do redukcji kwasów karboksylowych – kwas przeprowadzano w ester i ten dopiero poddawano redukcji z użyciem sodu w alkoholu.
Kondensacja Claisena
Reakcja kondensacji Claisena jest w wielu szczegółach analogiczna do kondensacji aldolowej. Podobnie jak tamta zachodzi w środowisku silnie zasadowym i rozpoczyna się od utworzenia karboanionu z cząsteczki estru:
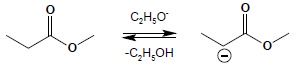
Utworzony karboanion (należy pamiętać, iż podobnie jak w przypadku kondensacji aldolowej, anion jest tworzony zawsze poprzez oderwanie protonu w pozycji α względem grupy karbonylowej estru, niezależnie od długości łańcucha), atakuje atom węgla grupy karbonylowej drugiej cząsteczki estru:
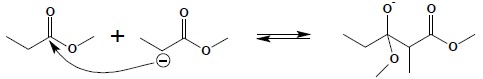
W następnym etapie, poprzez odłączenie anionu alkoholanowego tworzy się cząsteczka β-oksoestru. Reakcja jednak nie kończy się na tym etapie, ponieważ powstały β-oksoester tworzy sól w wyniku reakcji z silną zasadą obecną w mieszaninie reakcyjnej:
![]()
Otrzymanie końcowego produktu kondensacji, jakim jest β-oksoester, wymaga zakwaszenia mieszaniny reakcyjnej:
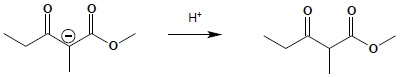
Ostatni etap reakcji kondensacji Claisena jest bardzo istotny z punktu widzenia mechanizmu reakcji. Poprzednie bowiem etapy są procesami równowagowymi, których stała równowagi jest silnie przesunięta w stronę substratów. Tworzenie anionu β-oksoestru, jako proces nieodwracalny, zaburza położenie równowagi poprzedzających etapów, powodując pojawianie się kolejnych porcji produktu. Konsekwencją istnienia tego etapu jest wymóg, aby ester poddawany kondensacji Claisena posiadał co najmniej dwa atomy wodoru w pozycji α względem grupy karbonylowej – pierwszy potrzebny jest do utworzenia anionu reagującego z drugą cząsteczką estru, drugi do utworzenia soli β-oksoestru.
Podobnie jak w przypadku kondensacji aldolowej możliwe jest przeprowadzenie krzyżowej (mieszanej) kondensacji Claisena. W tym przypadku, aby ograniczyć ilość możliwych produktów, substraty dobiera się, tak aby tylko jeden z nich posiadał atomy wodoru w pozycji α i używając tego estru w nadmiarze można z dobrymi wydajnościami otrzymać jeden produkt:
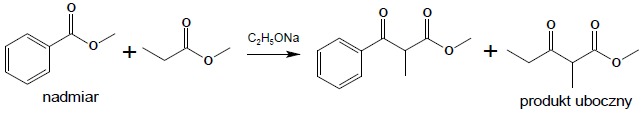
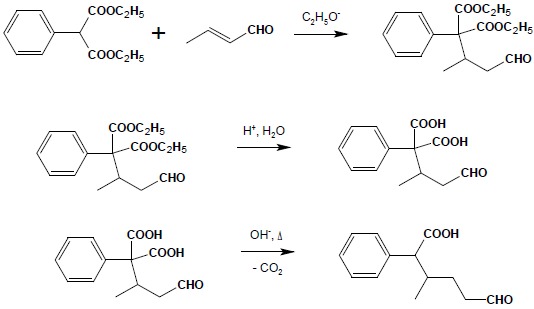
Reakcja Michaela
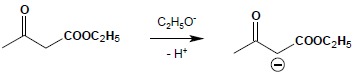
Jest to kolejny przykład reakcji addycji do α,β-nienasyconych związków karbonylowych. Dogodnymi substratami do tej reakcji są związki posiadające reaktywne atomy wodoru, czyli atomy wodoru grupy CH lub CH2 stojącej pomiędzy grupami karbonylowymi. Najczęściej wykorzystywanymi są estry kwasów malonowego i acetylooctowego oraz ich pochodnych. W pierwszym etapie reakcji cząsteczka estru reaguje z silną zasadą (alkoholan) – analogicznie rozpoczyna się reakcja kondensacji Claisena – tworząc karboanion:
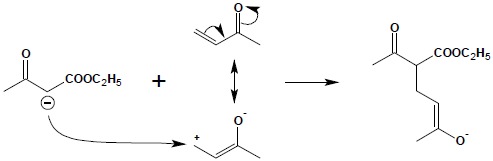
Powstały anion reaguje z nienasyconym związkiem karbonylowym, zgodnie z jego strukturą kanoniczną, obrazującą układ z rozdzielonym ładunkiem:
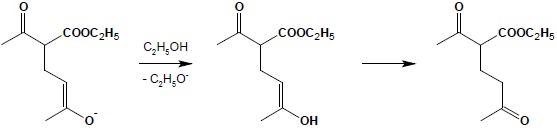
W ostatnim etapie następuje pobranie protonu z rozpuszczalnika (odtworzenie katalizatora) i przemiana tautomeryczna formy enolowej w ketonową:
Sumarycznie kondensacja Michaela może być rozpatrywana jako addycja 1,4 kwasu, jakim jest aktywna grupa CH związku 1,3-dikarbonylowego do α,β-nienasyconego związku karbonylowego. Reakcja ta stanowi bardzo dobrą metodę rozbudowy i łączenia różnych fragmentów węglowodorowych w większe struktury. W połączeniu z łatwą dekarboksylacją pochodnych kwasu malonowego czy β-oksokwasów kondensacja Michaela może być wykorzystywana do syntezy skomplikowanych kwasów lub ketonów. Poniższy przykład obrazuje zastosowanie kolejno reakcji kondensacji, hydrolizy grup estrowych produktu i dekarboksylacji w celu otrzymania złożonego kwasu monokarboksylowego:
Otrzymywanie estrów
Poniżej zebrano metody otrzymywania estrów:
a) bezpośrednia estryfikacja kwasów alkoholami (nie nadaje się do otrzymywania estrów fenoli)
b) reakcja chlorków i bezwodników kwasowych z alkoholami i fenolami
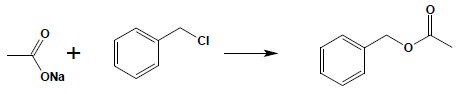
c) reakcja soli kwasów karboksylowych z halogenopochodnymi:
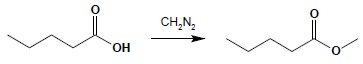
d) otrzymywanie estrów metylowych w reakcji kwasów z diazometanem: