
Onkogeneza (ang. oncogenesis), kancerogeneza (ang. carcinogenesis) lub inaczej nowotworzenie to długotrwały proces prowadzący do powstawania nowotworu. Związany jest on z nagromadzeniem się w komórce zmian genetycznych i epigenetycznych. W ich efekcie dochodzi do upośledzenia lub wzmocnienia ekspresji genów zaangażowanych w regulację cyklu komórkowego: protoonkogenów, genów supresorowych lub genów mutatorowych. Zmieniona komórka ulega niekontrolowanym podziałom i staje się niewrażliwa na sygnały kierujące ją na drogę samobójczej śmierci.
Nowotwór jako choroba genetyczna
Charakterystyczną cechą nowotworów jest ich zdolność do ulegania niekontrolowanym podziałom. W ich wyniku guz rozrasta się w obrębie tkanki macierzystej i niszczy otaczające go struktury, upośledzając przy tym czynności sąsiadujących narządów. Ta nieregularna mitoza prowadzi do heteroploidii, a znajdujące się w komórkach rakowych chromosomy często wykazują rearanżacje strukturalne.
Nowotwory są zatem chorobami o podłożu genetycznym i powstają w wyniku nagromadzenia się zmian w genomie już istniejących, zdrowych komórek. Teorię tą potwierdza fakt, że predyspozycje do zachorowań mogą być dziedziczone i w niektórych rodzinach występują ze wzmożoną częstotliwością.
Etapy kancerogenezy
Kancerogeneza jest procesem ciągłym, w którym można wyróżnić trzy podstawowe etapy:
• inicjację, czyli wystąpienie pierwszej, nieodwracalnej mutacji. Pierwotnie zmutowaną komórkę określa się jako komórkę macierzystą nowotworu. Charakteryzuje się ona trwałą zdolnością do samoodtwarzania i małą zdolnością do różnicowania się. Komórki macierzyste stanowią jedynie kilka procent wszystkich komórek tworzących masę guza. Reszta to tzw. komórki zejściowe nowotworu, które z czasem stają się niezdolne do dalszych podziałów;
• promocję, to znaczy nagromadzenie się zmian genetycznych oraz epigenetycznych prowadzących do konwersji zmutowanej komórki w komórkę nowotworową, tzn. ulegającą niekontrolowanym podziałom. Ten etap może trwać nawet przez kilka lat;
• progresję, podczas której nowotwór rozwija się, nabiera zdolności do naciekania tkanek oraz zdolności do przerzutowania (metastazy). Proces ten może trwać od kilku miesięcy do kilku lat.
Niekiedy wyróżnia się także etap preinicjacji, czyli narażenia na działanie czynników kancerogennych. Może on trwać całe życie i nie prowadzić do powstawania zmian nowotworowych.
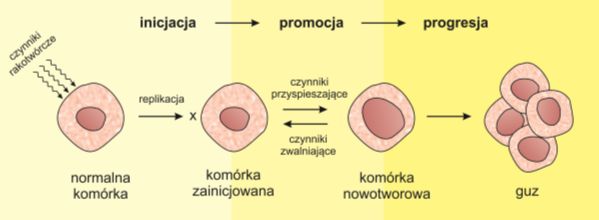
Rys. 1. Etapy kancerogenezy
Kancerogeny, czynniki rakotwórcze, onkogenne
Czynniki onkogenne są to czynniki, których działanie inicjuje procesy nowotworzenia. Można je podzielić na:
– czynniki chemiczne, np. niektóre metale ciężkie (nikiel, kadm, kobalt), substancje smoliste, azbest;
– czynniki fizyczne, np. promieniowanie UV i jonizujące;
– czynniki biologiczne: egzogenne takie jak wirusy (HPV), bakterie (Helicobacter pylori) oraz endogenne, np. błędy replikacji, pośrednie produkty przemiany materii (hormony, wolne rodniki).
Niestabilność genetyczna
Zwiększona podatność niektórych komórek na działanie kancerogenów związana jest z występowaniem tak zwanej niestabilności genetycznej. Cecha ta może się ujawniać jako:
1) niestabilność chromosomowa (ang. chromosomal instability, CIN), czyli nagromadzenie aberracji liczbowych (aneuploidie, poliploidie) oraz strukturalnych (np. translokacje, delecje, amplifikacje) chromosomów. Wśród aberracji chromosomowych wyróżnić można zmiany pierwotne, kluczowe dla rozwoju danego nowotworu, i wtórne, będące jednocześnie przyczyną i efektem niestabilności chromosomowej.
Specyficzne translokacje występują niemal w każdym typie nowotworu i często są wykorzystywane do odróżnienia pokrewnych typów tej choroby. Najlepiej scharakteryzowane pod względem translokacji są nowotwory układu krwiotwórczego, np. białaczki i chłoniaki.
Przykładowo chłoniak Burkitta (ang. Burkitt’s tumor) charakteryzuje się specyficzną translokacją 8 chromosomu z chromosomem 14, 2 lub 22. Najpowszechniej zachodzi translokacja t(8;14)(q24:q32), w wyniku której protonkogen myc znajduje się w bliskim sąsiedztwie sekwencji wzmacniającej, należącej do grupy genów ciężkiego łańcucha immunoglobulin. Wzmożona transkrypcja onkogenu stymuluje podziały komórkowe. Translokacje na chromosom 2 i 22 powodują przeniesienie protoonkogenu w pobliże genów lekkiego łańcucha immunoglobulin.
Translokacje mogą również przyczyniać się do powstawania nowych genów – onkogenów. Tworzą się one w wyniku połączenia końców 3’ i 5’ różnych genów. Najlepiej znanym przykładem jest chromosom Filadelfia (ang. Philadelphia chromosome, Ph) występujący w 95% przypadków przewlekłej białaczki szpikowej. Translokacja t(9;22)(q34;q11) prowadzi do połączenia dwóch protoonkogenów: bcr i abl. Mutacja blokuje naprawę DNA i wzmaga częstotliwość występowania podziałów komórkowych.
2) niestabilność mikrosatelitarna (ang. microsatellite instability, MIMSI), czyli zmiana długości alleli powstała w wyniku zwiększenia lub zmniejszenia liczby powtórzeń nukleotydowych.
Mikrosatelity są to równomiernie rozłożone, krótkie sekwencje tandemowe (ang. short tandem repeats, STR). Ich rola nie jest do końca poznana, wiadomo jednak, że rzadko występują w sekwencjach kodujących.
Niestabilność mikrosatelitarna powstaje w wyniku mutacji zachodzących w trakcie replikacji materiału genetycznego lub poreplikacyjnej naprawy DNA. Tego typu zmiana została po raz pierwszy odkryta w dziedzicznych, niepolipowatych rakach jelita grubego (ang. Hereditary Non-Polyposis Colorectal Cancer, HNPCC) i dziś stosuje się ją jako marker fenotypu mutatorowego RER+ (ang. Replication ERror phenotype). Wykazujące ją nowotwory określa się jako RER+.
Za pomocą analizy wybranych sekwencji mikrosatelitarnych można oznaczyć niestabilność alleliczną (ang. allelic instability), czyli inaczej utratę heterozygotyczności (ang. loss of heterozygosity; LOH) – utratę jednego z dwóch alleli tego samego genu. Zjawisko to występuje zarówno we wczesnych, jak i zaawansowanych stadiach procesu nowotworowego.

Rys. 2. Utrata heterozygotyczności w komórce nowotworowej. Delecja części chromosomu prowadzi do utraty jedynego aktywnego allelu genu supresorowego transformacji nowotworowej
4) niestabilność epigenetyczna, są to zmiany w ekspresji genów, niezwiązane bezpośrednio z sekwencją DNA. W odróżnieniu od zmian genetycznych, można je odwrócić poddając działaniu odpowiednich substancji chemicznych.
W przypadku nowotworów, niestabilność epigenetyczna dotyczy genów zaangażowanych w procesy różnicowania się i proliferacji komórek, regulację cyklu komórkowego, naprawę DNA, aktywność czynników wzrostu i apoptozę.
Określone zmiany epigenetyczne mogą być specyficzne dla danych typów nowotworów, np. glejaka wielopostaciowego oraz raka jelita grubego, stercza, płuc, przełyku, pęcherza moczowego.
Ich przyczyną są zaburzenia w rearanżacji struktury chromatyny lub nieprawidłowa metylacja DNA. Hipometylacja związana jest zazwyczaj z nadekspresją onkogenów, natomiast hipermetylacja wycisza funkcje genów supresowrowych.
Geny związane z onkogenezą
Zmiany odpowiedzialne za procesy nowotworzenia mogą zachodzić w:
• onkogenach,
• genach naprawy DNA (tzw. geny mutatorowe),
• antyonkogenach (genach supresorowych).
Onkogeny (ang. oncogenes)
Onkogeny są to geny, które ulegając ekspresji powodują przekształcenie się zdrowej komórki w nowotworową.
Prawidłowe wersje onkogenów nazywa się protoonkogenami. Są one obecne w każdym genomie i odpowiadają za właściwy przebieg cyklu komórkowego, a także kontrolują procesy różnicowania i apoptozy, czyli samobójczej śmierci.
Aktywacja protoonkogenu może się odbywać poprzez mutację lub amplifikację. W niektórych komórkach nowotworowych można znaleźć nawet kilka tysięcy kopii onkogenu, np. amplifikacja c-myc w neuroblastomie. Ich nadmierna aktywność i nadprodukcja kodowanych białek uczestniczących w szlakach sygnalnych, pobudza niekontrolowane podziały oraz zwiększa przeżywalność komórek. Oba te procesy przyczyniają się do powstawania guza.
Mutacje protoonkogenów mają charakter dominujący, tzn. już jedna kopia onkogenu wystarcza, aby spowodować zmiany w zachowaniu komórki.
Wyróżnia się:
1) onkogeny związane z czynnikami wzrostu, czyli peptydami, które za pomocą specyficznych receptorów stymulują wzrost i podziały komórek docelowych. Do tej grupy należą:
• onkogen sis – kodujący łańcuch β płytkowego czynnika wzrostu (ang. platelet derived growth factor, PDGF);
• onkogen fms – kodujący zmutowaną wersję czynnika 1 stymulującego wzrost kolonii (ang. colony stimulating factor-1, CSF-1);
• onkogen ras – kodujący białka z rodziny białek G, związane z błonami i przenoszące informacje z receptorów powierzchniowych do enzymów pośredniczących w powstawaniu tzw. przekaźników drugiego rzędu;
2) onkogeny jądrowe, czyli takie, które kodują czynniki transkrypcyjne regulujące ekspresję innych genów. Zalicza się do nich:
• gen myc – kodujący białko łączące się z określoną sekwencją DNA i pobudzające transkrypcję genów niezbędnych do podziału komórki;
• onkogeny jun i fos – kodujące podjednostki prawidłowych czynników transkrypcyjnych Jun i Fos tworzących czynnik AP-1;
• onkogen erbA kodujący skróconą wersję receptora dla hormonu tyroidowego.
Geny mutatorowe (ang. murator genes)
Geny mutatorowe zwane są również opiekunami genomu (ang. caretakers). Ich produkty są odpowiedzialne za utrzymanie integralności DNA i biorą udział w procesach naprawczych uszkodzonego materiału genetycznego.
Mutacje genów mutatorowych prowadzą do mikrosatelitarnej niestabilności genomu i warunkują zwiększoną predyspozycję do nowotworzenia. Do utraty ich funkcji konieczna jest inaktywacja obu alleli, mutacja ma zatem charakter recesywny.
Utrata funkcji pojedynczego allelu w komórkach germinalnych może stać się przyczyną powstawania dziedzicznych zespołów predyspozycji do rozwoju nowotworów.
Geny supresorowe transformacji nowotworowej (ang. tumor supressor genes)
W prawidłowych komórkach geny supresorowe transformacji nowotworowej funkcjonują jako negatywne regulatory cyklu komórkowego. Kodowane przez nie białka hamują wejście komórki w fazę mitozy, a także odgrywają ważną rolę w indukcji apoptozy. Geny te są zatem odpowiedzialne za kontrolę replikacji oraz za stabilność genetyczną komórki i związku z tym określa się je mianem strażników genomu (ang. gatekeepers).
Właściwości onkogennych nabierają one w wyniku mutacji powodujących utratę ich prawidłowej funkcji. Zmiany te mają charakter recesywny, tzn. muszą im ulec oba allele genu supresorowego. U chorych osób, najczęściej spotykana jest mutacja punktowa jednego allelu i delecja drugiego, choć sporadycznie obserwuje się także mutacje obu alleli. Utrata ekspresji przez geny supresorowe może zachodzić również w wyniku zjawisk epigenetycznych, takich jak modyfikacje chromatyny i nieprawidłowa metylacja DNA.
Uszkodzenie genów supresorowych transformacji nowotworowej staje się często przyczyną występowania dziedzicznych zespołów predyspozycji do rozwoju nowotworów. Przykładem może być siatkówczak (retinoblastoma), czyli nowotwór siatkówki oka, który warunkowany jest uszkodzeniem obu alleli genu supresorowego RB1. Gen ten koduje fosfoproteinę o wielkości 110 kDa, wiążącą DNA i hamującą transkrypcję protoonkogenów takich jak myc i fos. Retinobastoma występuje najczęściej w okresie wczesnego dzieciństwa u pacjentów, którzy odziedziczyli jeden zmutowany allel. Forma rodzinna, związana z przekazaniem recesywnego genu, obejmuje zwykle obie gałki oczne, natomiast sporadyczna, dotyczy najczęściej jednej gałki ocznej. W komórkach nowotworowych dochodzi do utraty heterozygotyczności, czyli delecji części chromosomu 13 zawierającej gen RB1.
Gen p53
Gen p53 jest zlokalizowany w krótszym ramieniu chromosomu 17. Jego mRNA ma wielkość 2,2 – 2,5 kb i koduje czynnik transkrypcyjny o masie 53kDa (stąd jego nazwa). W małych stężeniach białko to występuje we wszystkich typach komórek. Charakteryzuje się ono bardzo krótkim okresem półtrwania (6-2- minut) i jest zaangażowane w różne procesy komórkowe.
Stanowi ono m.in. główny punkt kontrolny cyklu komórkowego (ang. checkpoint), w którym komórki muszą się zatrzymać podczas przejścia z fazy G1 do fazy S, charakteryzującej się intensywną replikacją DNA. W przypadku wystąpienia uszkodzeń materiału genetycznego, następuje wzrost zawartości białka p53 w jądrze komórkowym. Zahamowanie podziałów umożliwia dokonanie naprawy zaistniałych błędów. Gdy straty są zbyt duże, p53 kieruje komórkę na drogę samobójczej śmierci. Białko to jest zatem strażnikiem genomu, który chroni komórkę przed uszkodzeniami mogącymi prowadzić do transformacji nowotworowej. Jego inaktywacja odbywa się na drodze fosforylacji. Może być ona również wynikiem mutacji, której efektem są niekontrolowane podziały komórkowe i zahamowanie apoptozy.
Mutacje genu p53 obserwuje się w ok. 50 % nowotworów człowieka. Wykazuje on przy tym właściwości zarówno onkogenów jak i genów supresorowych. W jego obrębie występuje wiele mutacji, takich jak zmiany punktowe, delecje i insercje. Zmutowane formy p53, wprowadzone razem z onkogenem ras do prawidłowych fibroblastów szczura, inicjują ich przemianę w komórki nowotworowe. W transformowanych komórkach transkrypt p53 wykazuje dłuższy okres półtrwania – od 4 do 8 godzin, co znacznie zwiększa ilość białka w komórce. W wielu nowotworach obserwuje się również delecje krótszego ramienia chromosomu 17, czyli zmiany charakterystyczne dla genów supresorowych transformacji nowotworowej.
Dzieje się tak dlatego, że p53 działa jako dimer. Połączenie się zmutowanej, tzn. nieaktywnej części z formą aktywną (niezmutowaną), prowadzi do powstania nieaktywnego kompleksu. Jest to tzw. efekt dominująco negatywny. Całkowita inaktywacja prawidłowego białka p53 następuje jednak dopiero w wyniku delecji części chromosomu zawierającej pozostały prawidłowy gen.
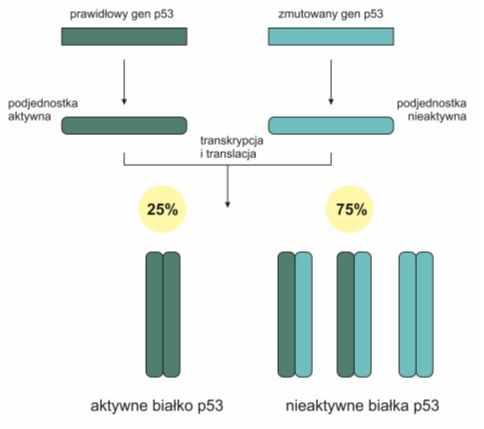
Rys. 3. Efekt dominująco negatywny zmutowanego genu p53. Białko zmutowane tworzy dimery z białkiem aktywnym i w ten sposób prowadzi do jego unieczynnienia
Rola wirusów w onkogenezie
Jedną z najważniejszych przyczyn powstawania raka u zwierząt są tzw. retrowirusy onkogenne. Retrowirusy są to wirusy RNA, których replikacja zachodzi przez intermediat, czyli DNA (prowirus) ingerujący z genomowym DNA gospodarza i ulegający transkrypcji do potomnego wirusowego RNA. Wirus wykorzystuje przy tym polimerazę RNA gospodarza.
Wiele retrowirusów onkogennych posiada dodatkowy gen, którego nie obserwuje się w spokrewnionych wirusach nieonkogennych. Onkogen ten różni się w zależności od typu wirusa i znajduje się pod kontrolą silnej sekwencji wzmacniającej, czyli enhancera zlokalizowanego w długich powtórzeniach końcowych (ang. long terminal repeat, LTR) genomu wirusa.
Nazwy onkogenów pochodzą zwykle od retrowirusów, u których zidentyfikowano je po raz pierwszy, np. v-myc – z ptasiego wirusa myleocytomatosis, c-neu w DNA wyekstrahowanym z guza tkanki nerwowej szczura. Onkogeny aktywowane bez udziału wirusa określa się mianem c-onc, natomiast onkogeny wirusowe: v-onc.
Dzięki zastosowaniu sond molekularnych do hybrydyzacji odkryto, że w komórkach prawidłowych znajdują się sekwencje DNA (protoonkogeny) homologiczne do onkogenów wirusowych. Pozwala to przypuszczać, że onkogeny retrowirusowe zostały przyłączone do genomu wirusowego włączonego w genom gospodarza (prowirusa) i pochodzą od protoonkogenów obecnych w prawidłowych komórkach.
Klasyfikacji wirusów jako tzw. bezpośrednich kancerogenów można dokonać w przypadku gdy:
• badania epidemiologiczne (w tym prospektywne) zidentyfikowały wirusa jako główny czynnik ryzyka nowotworu;
• wykazana została stała obecność genomu wirusa lub jego części w każdej komórce nowotworowej;
• transfekcja komórki za pomocą materiału genetycznego wirusa w warunkach in vitro lub w odpowiednim modelu zwierzęcym indukuje powstawanie nowotworu lub prowadzi do unieśmiertelnienia (ang. immortalization) komórki;
• usunięcie wirusowego materiału genetycznego z transfekowanych komórek lub zahamowanie funkcji wirusowego kwasu nukleinowego prowadzi do utraty przez komórkę cech immortalizacji lub złośliwego fenotypu;
• ryzyko wystąpienia nowotworu zmniejsza się w przypadku czynnej immunizacji w stosunku do wirusa. Obecnie istnieje możliwość zapobiegania wystąpieniu zmian przed- i nowotworowych za pomocą tzw. pierwotnej profilaktyki, czyli szczepieniom przeciw onkogennym wirusom hepatitis B oraz przeciw onkogennym typom HPV.
Na tej podstawie wyróżnić można trzy grupy wirusów o potencjale onkogennym:
• wirusy wykazujące ekspresję swoistych onkogenów niezbędnych do transformacji komórki, np. wirus Epsteina-Barr, onkogenny wirus HPV, ludzki wirus opryszczki typu 8 i ludzkie retrowirusy T-limfotropowe;
• wirusy z nabytymi komórkowymi onkogenami (tzw. acute transforming viruses), np. wirus mięsaka Rous’a i inne;
• wirusy posiadające zdolność do wbudowywania materiału genetycznego w swoiste miejsca w chromosomach gospodarza i aktywowania komórkowych onkogenów, np. wirus wywołujący guzy sutka u myszy.
Tab. 1. Związek różnych wirusów z nowotworami u człowieka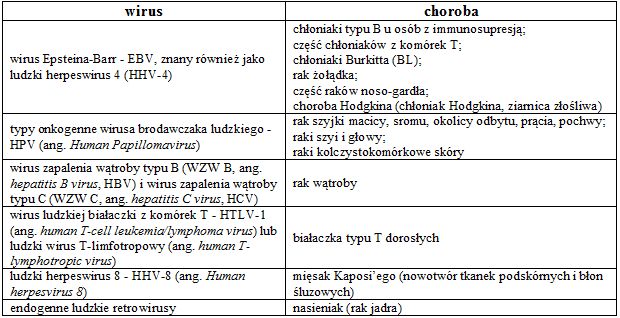
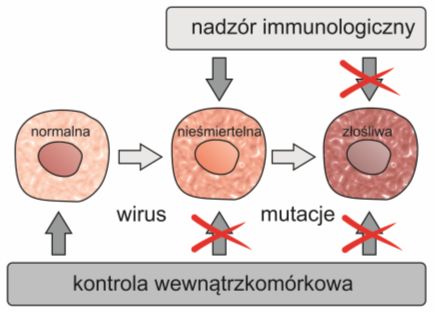
Rys. 4. Mechanizmy kontroli wewnątrzkomórkowej i zewnątrzkomórkowej w przebiegu ontogenezy wirusowej i progresji nowotworowej
Telomery i telomeraza w onkogenezie
Telomery są elementami strukturalnymi występującymi na końcach każdego chromosomu i chroniącymi je przed niehomologicznym łączeniem się końców i działaniem nukleaz. Ich obecność umożliwia także kompletną replikację nici opóźnionej.
Telomery nie kodują żadnych białek. Są one zbudowane z powtarzających się sekwencji TTAGGG połączonych białkami TBP (ang. telomere binding proteins) i tworzących strukturę składającą się z dwóch pętli D i T (ang. D-Loop i T-Loop). Długość telomeru waha się od 2 do 20 kpz i różni się w zależności od typu tkanki i jej wieku. W każdym cyklu komórkowym tracone jest około 50 -200 pz. Zbytnio skrócone telomery nie mogą dłużej pełnić swoich funkcji i w rezultacie komórka kierowana jest na drogę spoczynku lub apoptozy. Z tego powodu telomery określa się niekiedy mianem zegara molekularnego regulującego liczbę podziałów komórkowych.
Komórki które miały wejść w fazę spoczynku, mogą ją ominąć w obecności wirusowych onkogenów takich jak np. SVA40 duży antygen T (ang. large T antygen, LT) oraz HPV białka E6 i E7. Białka te mogą inaktywować p53 oraz Rb, czyli czynniki supresji nowotworów, pełniące kluczową rolę podczas kierowania komórek na drogę spoczynku. Mechanizm ten polega najprawdopodobniej na regulacji telomerazy stanowiącej integralną część każdego telomeru. Enzym ten jest polimerazą DNA zależną od RNA, która wykorzystuje własny RNA jako matrycę do wydłużania 3’ końca, tzn. syntetyzowania charakterystycznych powtórzeń telomeru. Stosując metodę TRAP (ang. telomeric repeat amplification protocol) wykazano, że aż 85-90% typów nowotworów posiada aktywną telomerazę.
Samo unieśmiertelnienie komórki nie jest równoznaczne z jej przekształceniem w komórkę nowotworową. Telomeraza jest również aktywna m.in. w zdrowych komórkach embrionalnych oraz w komórkach somatycznych zdolnych do odnawiania się tzn. komórkach macierzystych skóry, hemopoetycznych komórkach macierzystych, limfocytach oraz komórkach krypt jelitowych. Niemniej jednak tendencja do nadmiernych podziałów może prowadzić do nagromadzenia się mutacji istotnych dla zainicjowania procesów nowotworzenia.
Należy również zaznaczyć, że długość telomeru w komórkach nowotworowych jest znacznie mniejsza niż w komórkach prawidłowych i dlatego potrzebują one stałej aktywności telomerazy do procesów podziału. Brak enzymu kieruje komórki nowotworowe na drogę apoptozy. Zablokowanie aktywności telomerazy może zatem zahamować procesy nowotworzenia bez większej szkody dla zdrowych komórek. Efekt ten uzyskuje się dzięki immunoterapii, terapii genowej, inhibitorom odwrotnej transkryptazy, stabilizacji kwadrodupleksów oraz blokowaniu dostępu telomerazy do telomerów.
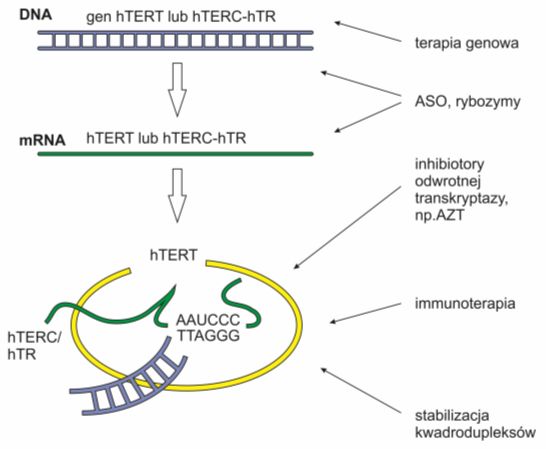
Rys. 5. Podstawowe metody blokowania telomerazy
Autor: Anna Kurcek
Literatura:
1. Feldman R., Nowotwory od podstaw.
2. Jesionek-Kupnicka D., Jabłońska J., 2004. Zmiany epigenetyczne w nowotworach. Onkol. Pol., 7(40): 181-185.
3. Kowalska A., Kowalik A., 2006. Telomer i telomeraza w ontogenezie. Współczesna Onkologia, 10(10): 485–496.
4. Kozłowska J., Łaczmańska I., 2010. Niestabilność genetyczna – jej znaczenie w procesie powstawania nowotworów oraz diagnostyka laboratoryjna. Journal of Oncology, 60(6): 548–553.
5. Łaczmańska I., Kozłowska J., 2009. Niestabilność chromosomowa w sporadycznym raku jelita grubego. Współczesna Onkologia, 13(4): 177-180.
6. Majewski S., 2007. Rola wirusów w onkogenezie. Alergia, 2: 24-27.
7. Mu J., Wei L.X., 2002. Telomere and telomerase in oncology. Cell Research, 12: 1–7.
8. Śmigiel R., Stembalska A., Stal A., Jonkisz A., Trusewicz A., Dobosz T., Grzebieniak Z., Sąsiadek M., 2006. Badania niestabilności mikrosatelitarnej u pacjentów chorych na raka jelita grubego leczonych na Dolnym Śląsku. Adv Clin Exp Med, 15 (1): 29–36.
9. Turner P.C., McLennan A.G., Bates A.D., White M.R.H., 2004. Krótkie wykłady – Biologia Molekularna. Wydawnictwo Naukowe PWN.
10. Urbanowicz I., Stacherzak-Pawlik J., Woźniak M., 2004. Aktywność telomerazy i długość telomerów w rozrostach układu chłonnego. Medycyna Rodzinna, 4: 187-192.
11. Winter P.C., Hickey G.I., Fletcher H.L., 2004. Krótkie wykłady – Genetyka. Wydawnictwo Naukowe PWN.