
PRZEDRUK, oryginał dostępny pod adresem www
Tytuł oryginalny: Badanie kinetyki enzymów
Uniwersytet Gdański (www)
Wydział Chemii (www)
Katedry Chemii Bioorganicznej (www)
Kierownik: Prof. dr hab. Krzysztof Rolka
Adres:
ul. Sobieskiego 18/19
80-952 Gdańsk
Kontakt: tel. 58 52 35 386
____________________________________________________________________________
Przy stałym stężeniu enzymu, a przy zmieniającym się początkowym stężeniu substratu, zmiany szybkości reakcji katalizy, wyrażonej jako liczba moli substratu przetworzonego w jednostce czasu, można przedstawić w postaci krzywej hiperbolicznej (rys. 1).
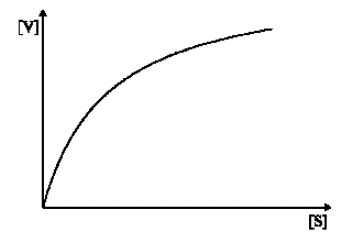
Rys. 1 Zależność szybkości reakcji enzymatycznej w funkcji stężenia substratu
Przy małych stężeniach substratu niektóre cząsteczki enzymu nie tworzą kompleksu z substratem, co powoduje, że enzym nie wykazuje swojej maksymalnej aktywności katalitycznej. Maksymalną szybkość reakcji osiąga dopiero przy wyższym stężeniu substratu, kiedy wszystkie cząsteczki enzymu będą tworzyć kompleks enzym – substrat. Całkowite wysycenie enzymu substratem powoduje, że dalsze zwiększanie stężenia substratu nie może już wpłynąć na zwiększenie szybkości reakcji: reakcja przebiega ze stałą, maksymalną szybkością.
Fakt ten tłumaczy się, zgodnie z modelem zaproponowanym w 1913 roku przez L. Michaelisa i M. Menten, powstawaniem kompleksu enzym – substrat w trakcie katalizy enzymatycznej. Najprostszy model, który tłumaczy kinetykę wielu enzymów, wygląda następująco:
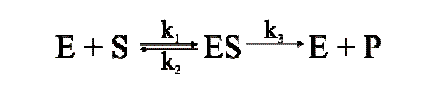
Równanie 1.
Enzym (E) łączy się z substratem (S) tworząc kompleks ES; reakcję charakteryzuje stała szybkości k1. Istnieją dwie możliwe drogi rozpadu kompleksu ES. Może on dysocjować do E i S ze stałą szybkości k2 lub może się przekształcać, tworząc produkt (P) ze stałą szybkości k3. Reakcje enzymatyczne stanowią zwykle złożony układ, składający się z kilku pośrednich reakcji o różnych stałych szybkości k. Szybkości poszczególnych przemian (równanie 1) przedstawiają się następująco:
V1 = k1×[E]×[S] (2)
V2 = k2×[ES] (3)
V3 = k3×[ES] (4)
Birgs i Halden w 1925 roku po raz pierwszy wprowadzili założenie występowania stanu stacjonarnego w przebiegu reakcji enzymatycznej. Badacze ci założyli, że istnieje taki moment reakcji, w którym szybkość tworzenia kompleksu ES równa się sumie szybkości jego rozpadu na produkt (P) i substrat (S), co jest równoważne z warunkiem niezmienności stężenia kompleksu ES. Kompleks ES tworzy się z szybkością V1, a rozpada z szybkością V2 i V3. W stanie równowagi można przyjąć, że:
V1 = V2 + V3.
Stąd: k1×[E]×[S] = (k2+k3)×[ES] (5)
Szybkość początkowa reakcji zależy od szybkości rozpadu kompleksu ES (k2+k3), od stężenia kompleksu oraz szybkości jego powstawania (k1) i dlatego stała równowagi takiego układu wyraża się wzorem (6) i nosi nazwę stałej Michaelisa (KM).
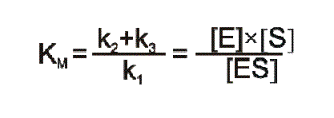
Wzór 6. Stała Michaelisa
Stężenie enzymu (E) w powyższym wzorze jest stężeniem enzymu wolnego, niezwiązanego w kompleksie, czyli równe początkowemu stężeniu enzymu (Ep) pomniejszonemu o stężenie kompleksu ES. Można więc napisać:
[E] = [Ep]-[ES] (7)
Szybkość reakcji enzymatycznej zależy głównie od stężenia kompleksu ES i szybkości tworzenia się z niego produktu i wynosi:
V = V3 = k3×[ES] (8)
Przekształcając i podstawiając odpowiednio równania (7) i (8) do równania (6) otrzymuje się:
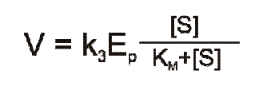
gdzie: k3 = kcat, zdefiniowane jako stała katalityczna
Maksymalną szybkość reakcji Vmax uzyskuje się wtedy, kiedy wszystkie miejsca enzymu są wysycane przez substrat, to znaczy, kiedy [S] >> KM, czyli [S] / ([S]+KM) jest bliskie 1.
Stąd: Vmax = k3 × Ep (10)
W wyniku podstawienia równania (10) do równania (9) otrzymuje się równanie Michaelisa-Menten:
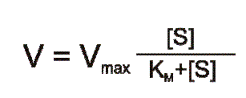
Rozwiązaniem równania (11) jest krzywa przedstawiona na rysunku 2.
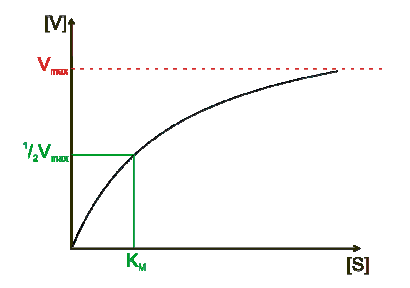
Rys. 2 Wykres zależności szybkości reakcji V od stężenia substratu [S], dla enzymu zachowującego się zgodnie z kinetyką Michaelisa-Menten
Równanie Lineweavera-Burka (12) jest przekształceniem wzoru Michaelisa Menten:
1/V = KM/Vmax x 1/[S] + 1/Vmax
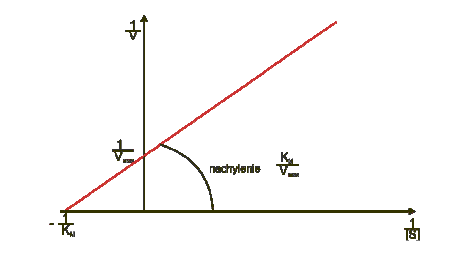
Rys. 3. Krzywa Lineweavera-Burka
Przekształcenie to ma na celu uzyskanie równania o ogólnej postaci linii prostej (y = ax + b).
Wartość KM / Vmax (a) i 1/Vmax (b) są wielkościami stałymi, stąd zależność 1/V (y) od 1/[S] (x) jest linią prostą, której nachylenie określa zawsze stosunek KM/Vmax (współczynnik regresji a) i która przecina oś y (czyli 1/V) w punkcie wyznaczającym 1/Vmax.
Zadaniem niniejszego doświadczenia będzie wyznaczenie parametrów kinetycznych (Vmax, kcat = Vmax/E0, KM i kcat/KM) w układzie enzym – substrat. Stosowana w doświadczeniu metoda polega na spektrofotometrycznym pomiarze szybkości hydrolizy substratu (p-nitroanilidu-D, Largininy), której miarą jest przyrost absorbancji uwalnianego chromoforu (p-nitroaniliny).
Odczynniki i sprzęt laboratoryjny:
1. Spektrofotometr UV-Vis
2. Kuwety plastikowe
3. Roztwór trypsyny (7 mg/mL 1 mM HCl i 20 mM CaCl2), (pH 3,0)
4. Roztwór substratu chromogenicznego (p-nitroanilidu-D, L-argininy) (stężenie roztworu podstawowego 10 mg/mL DMSO)
5. Bufor weronalowy o stężeniu 0,1 M z dodatkiem 20 mM CaCl2, (pH 8,3)
6. Roztwór NPGB (p-guanidynobenzoesan p’-nitrofenylu) (0,84 mg związku rozpuszczonego w mieszaninie złożonej z 400 µL acetonitrylu i 100 µL DMF)
7. Bufor 0,1 M Tris/HCl zawierający 20 mM CaCl2 oraz 0,005% Tritonu X-100, pH 8,3
8. Pipety miarowe automatyczne 20-200 µL i 500-5000 µL
Wykonanie doświadczenia:
Metodyka wyznaczania parametrów kinetycznych w układzie enzym – substrat. Oznaczenie parametrów kinetycznych substratów chromogenicznych obejmuje następujące etapy:
1. Wyznaczenie stężenia aktywnej formy enzymu.
2. Wyznaczenie stężenia roztworów substratów.
3. Wyznaczenie parametrów kinetycznych.
Wyznaczenie stężenia aktywnej formy enzymu
Wyznaczanie stężenia bydlęcej β-trypsyny przeprowadza się poprzez miareczkowanie jego centrów aktywnych substratem wybuchowym (ang. burst kinetics substrate), jakim jest NPGB (p-guanidynobenzoesan p’-nitrofenylu). Wprowadzenie trypsyny do buforu zawierającego NPGB, powoduje gwałtowny przyrost absorbancji („wybuch barwy”). Podczas reakcji hydrolizy katalizowanej przez bydlęcą β-trypsynę, uwolniony zostaje p-nitrofenol (barwa żółta), którego absorbancja mierzona przy długości fali λab = 410 nm jest proporcjonalna do stężenia enzymu.
Miareczkowanie roztworu bydlęcej β-trypsyny przeprowadza się w jednorazowych kuwetach polistyrenowych, zawierających 1,5 mL buforu weronalowego o stężeniu 0,1 M z dodatkiem 20 mM CaCl2, (pH 8,3) . Roztwór podstawowy enzymu otrzymać przez rozpuszczenie 7 mg enzymu w 1 mL roztworu o składzie: 1 mM HCl i 20 mM CaCl2, (pH 3,0). Roztwór NPGB otrzymać przez rozpuszczenie 0,84 mg związku w mieszaninie złożonej z 400 µL acetonitrylu i 100 µL DMF.
Następnie do 5 kuwet pomiarowych dodawać 20 µL NPGB, a następnie 50 µL roztworu trypsyny. W kolejnym etapie dokonać pomiaru przyrostu absorbancji p-nitrofenolu przy długości fali λab = 410 nm. Po zakończeniu pomiarów obliczyć stężenie trypsyny jako średnią wartość, pochodzącą z wykonanych prób według wzoru:
C = (ΔA/16595) x (1570/50) [mol/dm3]
Przy obliczeniach uwzględnić rozcieńczenie i przyjąć wartość molowego współczynnika absorpcji p-nitrofenolu jako 16595 dm3×mol-1×cm-1.
Standaryzacja roztworu substratu
10 mg substratu chromogenicznego (p-nitroanilidu-D,L-argininy) rozpuścić w 1 mL dimetylosulfotlenku (DMSO). Następnie rozcieńczyć roztwór podstawowy 2, 5, 10 i 20 razy. Z każdego rozcieńczonego roztworu substratu pobrać 20 µL i dodać do jednorazowej kuwety polistyrenowej zawierającej 1,5 mL buforu 0,1 M Tris/HCl zawierającego 20 mM CaCl2 oraz 0,005% Tritonu X-100, pH 8,3. W kolejnym etapie do kuwety dodać 20 µL roztworu podstawowego trypsyny. Reakcję hydrolizy prowadzić do momentu uzyskania stałej wartości absorbancji przy λab = 410 nm, zwykle kilkanaście minut. Stężenie substratu obliczyć zgodnie z prawem absorpcji Lamberta-Beera, stosując molowy współczynnik absorpcji dla p-nitroaniliny wynoszący 9800 M-1 × cm-1.
Procedura wyznaczania parametrów kinetycznych
Do kilkudziesięciu jednorazowych kuwet polistyrenowych i zawierających 1,5 mL buforu 0,1 M Tris/HCl zawierającego 20 mM CaCl2 oraz 0,005% Tritonu X-100, pH 8,3, dodać po 20 µL roztworu substratu o odpowiednim stężeniu, a następnie 20 µL roztworu enzymu o dobranym wcześniej stężeniu (stężenie podane przez prowadzącego ćwiczenia). Objętość dodawanego substratu i enzymu jest zawsze stała i wynosi 20 µL.
Kuwetę pomiarową umieścić w spektrofotometrze i zarejestrować wartość absorbancji w następujących odstępach czasu: 0 sekund, 60 sekund, 120 sekund i 180 sekund. Na podstawie zmierzonych wartości absorbancji wyznaczyć w oparciu o równanie regresji liniowej początkowe szybkość hydrolizy substratu (rys. 4).
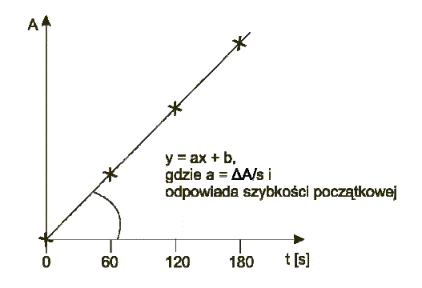
Rys. 4. Wyznaczanie początkowych szybkości hydrolizy (V)
Otrzymane wartości początkowych szybkości hydrolizy (V) wyznaczone przy zmieniającym się stężeniu substratu, wyrażone w jednostkach absorbancji na minutę (A/s), korzystając z prawa Lamberta Beera, przeliczyć na wartości początkowych szybkości hydrolizy wyrażone w mol/s. Dla wyznaczonych wartości V i [S] obliczyć 1/V oraz 1/[S] i wyniki umieścić w tabeli:
Na podstawie danych zawartych w tabeli narysować krzywą Lineweavera-Burka stanowiącą graficzne rozwiązanie równania (12). Wyznaczyć równanie regresji liniowej, gdzie;
a = KM/Vmax i b = 1/Vmax,
a następnie obliczyć Vmax i KM, oraz znając stężenie enzymu (E0), obliczyć wartość:
kcat = Vmax/E0 oraz wartość stałej specyficzności kcat/KM.