
Onkogeny są to geny, które ulegając ekspresji powodują przekształcenie się prawidłowej komórki w nowotworową. Ich prawidłowe wersje, czyli protoonkogeny występują powszechnie w organizmach zwierzęcych i pełnią funkcje niezbędne do zachowania integralności tkanek i organów. Między innymi kontrolują podziały komórkowe i procesy różnicowania, a także hamują samobójczą śmierć, czyli apoptozę. Zmutowane protoonkogeny ulegają nadmiernej ekspresji, a przez to stymulują namnażanie się komórek. Prowadzi to do utworzenia się guza i rozwoju nowotworu. Obecnie onkogeny stanowią jeden z głównych celów molekularnych leków przeciwnowotworowych.
Genetyczne podłoże nowotworów
Do uformowania się nowotworu dochodzi w momencie, kiedy organizm traci kontrolę nad podziałami i różnicowaniem się własnych komórek. Przyczyną są zmiany o charakterze genetycznym, czyli mutacje. Zachodzą one w obrębie charakterystycznych genów zaangażowanych w przebieg cyklu komórkowego, to znaczy onkogenów i genów supresorowych nowotworu, zwanych inaczej antyonkogenami. Ich wynikiem jest nieregularna mitoza i niewrażliwość na apoptozę. Cechy te prowadzą do uformowania się guza, który rozrastając się niszczy otaczające go tkanki i organy. Dodatkowe upośledzenie funkcji naprawczych jest przyczyną heteroploidii i akumulacji zmian w strukturalnej aranżacji chromosomów.
Komórki rakowe dzielą się i rosną bardzo szybko. Nie radzą sobie z dodatkowymi chromosomami i w związku z tym, podczas kolejnych cykli komórkowych, dochodzi do nieprawidłowej segregacji materiału genetycznego i nagromadzenia się dalszych mutacji. Zjawisko to określa się mianem niestabilności genetycznej.
Teorię o genetycznym podłożu nowotworów potwierdza fakt, że predyspozycje do zachorowań mogą być dziedziczone. W niektórych rodzinach obserwuje się wysoką częstotliwość nawet rzadkich form raka.
Pierwsze odkrycia
Na początku dwudziestego wieku, amerykański naukowiec Francis Peyton Rous prowadził badania nad mechanizmem przenoszeniem mięsaka między kurczakami. Sporządzał on płynne ekstrakty z wycinków guzów i aplikował je zdrowym zwierzętom. Zdołał w ten sposób wywołać u nich śmiertelną chorobę. Przygotowywane wyciągi przepuszczał przez filtry na tyle drobne, aby uniemożliwiły przedostawanie się bakterii. Były one jednak na tyle duże, że nie blokowały przejścia wirusom. W ten sposób odkryto pierwszego onkowirusa, czyli wirusa powodującego powstawanie guzów. Na cześć naukowca nazwano go wirusem mięsaka Rousa (ang. Rous sarcoma virus, RSV).
Dalsze badania wykazały, że RSV jest retrowirusem, czyli wirusem RNA przeprowadzającym proces odwrotnej transkrypcji. Właściwości onkogennych nabył on najprawdopodobniej w wyniku błędu, to znaczy przenoszony przez niego onkogen został wbudowany w jego genom przypadkowo, podczas procesów replikacji jakim ulegał w organizmie poprzedniego gospodarza. Pochodzący od organizmów wyższych protoonkogen nazwano c-src. Udowodniono, że występuje on w genomach wielu różnych gatunków i jest zaangażowany w regulację podziałów komórkowych i procesów wzrostowych. Również ludzie posiadają własną wersję genu c-src, a jego wysoka ekspresja towarzyszy różnym typom nowotworów. W wyniku infekcji wirusowej sekwencja tego genu może być zmieniona w taki sposób, aby kodowane przez niego białko wykazywało nietypową aktywność, np. może się on dostać pod kontrolę silnych promotorów i enhancerów zlokalizowanych w genomie wirusa. Utworzony w ten sposób onkogen v-src prowadzi do niekontrolowanego wzrostu i podziałów komórek i staje się przyczyną powstawania guza. Za to przełomowe odkrycie Michael Bishop i Harold Varmus otrzymali w 1975 roku nagrodę Nobla.
Onkogeny
Każda zdrowa komórka ma w swoim zestawie genów tzw. protoonkogeny. Kodowane przez nie białka uczestniczą w szlakach przekazywania sygnałów i odgrywają kluczową rolę w komórkowych mechanizmach wzrostu, dojrzewania i różnicowania. Niektóre z nich są również zaangażowane w regulację procesów samobójczej śmierci komórkowej.
W wyniku mutacji protoonkogeny mogą się przekształcać w tzw. onkogeny, czyli geny wykazujące nadmierną aktywność. Onkogen, w przeciwieństwie do protoonkogenu, jest niewrażliwy na bodźce, które powodują jego wyłączenie. Najprościej mówiąc: działa, choć już nie powinien. Steruje, już bez potrzeby, dalszą produkcją określonych białek, np. czynników wzrostu, czy też receptorów dla tych czynników, czego końcowym efektem są istotne zaburzenia wzrostu i różnicowania się komórek.
Ponadto onkogeny mają charakter dominujący w stosunku do protoonkogenów. Oznacza to, że zaledwie jedna ich kopia wystarcza, aby spowodować niekorzystne zmiany, takie jak niewrażliwość na apoptozę, ciągły wzrost i zdolność do ulegania niekontrolowanym podziałom komórkowym. W efekcie tworzy się guz, czyli nowotwór.
Protoonkogen a onkogen
Mutacje protoonkogenów powodują zmiany w ich ekspresji lub aktywności. Protoonkogeny i onkogeny mogą zatem różnić się od siebie:
• ilościowo, kiedy pierwotne funkcje genu pozostają niezmienione, a szkodliwe działanie warunkowane jest ich nadmierną ekspresją, np. gdy znajdą się pod kontrolą wirusowego promotora lub sekwencji wzmacniającej, lub też zostaną przeniesione do nowego miejsca w genomie. Wzmożona transkrypcja prowadzi do nadmiernego nagromadzenia się prawidłowych produktów genu.
Przykładem może być rak sutka u myszy wywoływany przez mysi retrowirus raka sutka (ang. Mouse mammary tumor virus, MMTV). Wbudowanie prowirusa MMTV w pobliże mysiego genu int-2 kodującego czynnik wzrostu, wzmaga ekspresję tego protoonkogenu i warunkuje nadmierne podziały komórkowe.
• jakościowo, to znaczy, gdy sekwencja kodująca ulega zmianom, np. delecjom lub mutacjom punktowym, a powstający produkt jest odmienny funkcjonalnie od prawidłowego białka i najczęściej cechuje się hiperaktywnością.
Przykładowo w wyniku integracji wirusa białaczki ptasiej (ang. Avian Leukemia Virus, ALV) w obrębie protoonkogenu erbB dochodzi do tworzenia receptora dla naskórkowego czynnika wzrostu (ang. Epidermal Growth Factor Receptor, EGFR), który nie posiada części zewnątrzkomórkowej wiążącej ligand. W efekcie tej zmiany receptor traci wrażliwość na EGF i ulega stałej aktywacji stymulując nieprzerwanie podziały komórkowe.
Aktywacja onkogenów
Istnieje kilka genetycznych mechanizmów mogących prowadzić do nadmiernego pobudzenia onkogenów:
• mutacje punktowe, delecje lub insercje, w których wyniku tworzą się produkty o wyższej niż zwykle aktywności, np. onkogen ras z mutacją punktową hamującą aktywność GTP-azy;
• mutacje punktowe, delecje lub insercje regulujące regiony promotorowe protoonkogenu i warunkujące jego nadmierną transkrypcję, np. czynnik AP-1 kodowany przez fos i jun;
• amplifikacje genów prowadzące do utworzenia dodatkowych kopii protoonkogenu. W niektórych komórkach nowotworowych znaleźć można nawet kilka tysięcy kopii onkogenu, czego przykładem jest amplifikacja c-myc w neuroblastomie (nerkowiaku);
• translokacje chromosomowe, w wyniku których protoonkogeny zostają przenoszone w miejsca warunkujące ich nadmierną ekspresję, np. ulokowanie protoonkogenu w regionie promotora innego genu, np. gen c-myc przenoszony w pobliże genu kodującego ciężki łańcuch immunoglobulin;
• translokacje chromosomowe które prowadzą do fuzji pomiędzy protoonkogenem a innym genem, a utworzone w ten sposób produkty fuzyjne wykazują o onkogennej aktywności. Najlepiej znanym przykładem tego typu onkogennej chromosomowej translokacji jest chromosom Filadelfia odkryty w 1960 roku i związany m.in. z przewlekłą białaczka szpikową (ang. myelogenous leukemia, CML). Powstaje on w wyniku połączenia się części chromosomu 9 z fragmentem chromosomu 22. Na złamanym końcu chromosomu 22 zlokalizowany jest gen BCR, który ulega fuzji z genem ABL1 z przyłączonego fragmentu chromosomu 9. Utworzony w ten sposób gen fuzyjny ulega ekspresji i koduje białko wykazujące wysoką aktywność kinazy tyrozynowej. Jego niekontrolowana ekspresja aktywuje inne białka zaangażowane w regulację cyklu komórkowego.
Tab.1 Przykłady onkogenów aktywowanych przez translokację.![]()
Podział onkogenów
Najczęściej onkogeny dzieli się ze względu na pełnione funkcje. W ten sposób można wśród nich wyróżnić:
• czynniki wzrostu, czyli specyficzne peptydy, które za pośrednictwem swoistych receptorów stymulują wzrost i podziały komórek docelowych
Onkogen sis – koduje łańcuch β płytkowego czynnika wzrostu (ang. platelet derived growth factor, PDGF), czyli jednego z głównych mitogenów (czynników indukujących mitozę) znajdujący się w krwi pełnej. Obecność onkogenu warunkuje autokrynną stymulację komórek nowotworowych oraz parakrynną symulację komórek sąsiadującego z guzem podścieliska i układu naczyniowego. Autostymulacja wzrostu komórek nowotworowych jest możliwa dzięki temu, że są one zaopatrzone w odpowiednie receptory powierzchniowe złożone z części zewnątrzkomórkowej i wewnątrzkomórkowej, czyli kinazy tyrozynowej podzielonej na dwie części katalityczne. Przyłączenie właściwego liganda warunkuje internalizację kompleksu i fosforylację tyrozyny, która w efekcie tworzy miejsce dla licznych wewnątrzkomórkowych substratów aktywujących kinazę PDGFR. W ten sposób dochodzi do pobudzenia ekspresji odpowiednich genów zaangażowanych we wzrost i podziały komórki. Kinaza może również aktywować i inaktywować cząsteczki biorące udział w procesie samobójczej śmierci.
Zwiększona ekspresja PDGF oraz jego receptora występuje m.in. w ludzkim niedrobnokomórkowym raku płuca.
• receptory czynników wzrostu
Wiele protoonkogenów koduje powierzchniowe receptory, których rola polega na przekazywaniu do wnętrza komórki sygnałów pochodzących ze środowiska zewnętrznego. Receptory takie składają się zazwyczaj z trzech funkcjonalnych części:
– regionu zewnątrzkomórkowego, wyeksponowanego na zewnątrz komórki i pełniącego rolę anteny zbierającej sygnały pochodzące z otaczającego ją środowiska;
– regionu wewnątrzbłonowego, pełniącego funkcję łącznika pomiędzy częścią zewnętrzną i wewnętrzną;
– regionu wewnętrznego, który często wykazuje swoją własną aktywność enzymatyczną i może łączyć się z innymi białkami zlokalizowanymi we cytoplazmie. W ten sposób aktywuje odpowiednie szlaki sygnałowe warunkujące właściwą odpowiedź komórki na zmiany zachodzące w środowisku zewnętrznym.
Zewnętrzne regiony łączą się z właściwymi dla siebie ligandami, np. czynnikami wzrostu lub czynnikami angiogenezy. Związanie liganda zmienia konformację receptora, co z kolei prowadzi do aktywacji domeny wewnątrzkomórkowej i wywołuje kaskady reakcji stymulujących wzrost komórki, jej proliferację lub śmierć.
Onkogeny mogą być związane z działaniem takich receptorów jak EGFR, czyli receptor nabłonkowego czynnika wzrostu (ang. epidermal growth factor; EGF) stymulujący wzrost komórek, czy też KDR – receptor dla czynnika wzrostu śródbłonka naczyniowego (ang. vascular endothelial growth factor; VEGF) zaangażowanego w procesy angiogenezy.
Innym przykładem jest onkogen erbA, który razem z erbB odkryto w ptasim wirusie erytroblastozy – AEV. ErbA koduje skróconą wersję receptora dla hormonu tyroidowego. W normalnych warunkach receptory te działają jako czynniki transkrypcyjne i w odpowiedzi na przyłączenie hormonu regulują ekspresję określonego genu. Białko ErbA nie posiada karboksylowego końcowego regionu występującego w prawidłowym receptorze i dlatego nie może przyłączyć hormonu, a co za tym idzie stymulować transkrypcji genu. Może jednak nadal łączyć się z tym samym miejscem w DNA i w związku z tym działa jak antagonista prawidłowego receptora hormonu tyroidowego.
• przekaźniki sygnałów, czyli czynniki które przekazują informacje z receptorów powierzchniowych do enzymów pośredniczących w powstawaniu tzw. przekaźników drugiego rzędu
Przykładem może być onkogen ras. Białka Ras poznano jako produkty genów retrowirusów onkogennych, a następnie jako onkogeny komórkowe. Są to białka o aktywności GTPazowej, podobnje do białek G. W stanie aktywnym wiążą guanozynotrifosforan (ang. guanosine-5′-triphosphate, GTP), natomiast w stanie nieaktywnym guanozynodifosforan (ang. guanosine diphosphate, GDP).
Białka typu Ras tworzą nadrodzinę złożoną z sześciu rodzin: Ras, Rho, Rab, Arf, Sar i Ran. Są one częścią wieloszczeblowej kaskady molekularnych interakcji i uczestniczą w przekazywaniu do jądra komórkowego sygnałów wzrostowych pochodzących z receptorów błony plazmatycznej. Sygnały te wywołują kaskadę reakcji wzmagających mitogenezę, to znaczy odpowiednią organizację cytoszkieletu, syntezę DNA oraz metabolizm lipidów.
Pochodząca z receptora wiadomość jest przekazywana na białko Ras za pośrednictwem czynnika wymiany nukleotydu guaniny (ang. guanine nucleotie exchange factors, GEF), zamieniającego zwiazaą z Ras cząsteczkę GDP na GTP. Zmiana ta powoduje subtelną rearanżację białka, które dzięki temu wywołuje kaskadę kinaz białkowych fosforylujących np. czynniki transkrypcyjne dostarczające informację do celu przeznaczenia. Liczba aktywnych Ras jest ściśle związana z ilością receptorów powierzchniowych, a wysyłany przez nie sygnał podlega autoregulacji. Za pomocą białka aktywującego GTP-azę (ang. GTPase-activating proteins, GAP) Ras hydrolizuje GTP do GDP i w ten sposób sam blokuje pełnione przez siebie funkcje.
Onkogen ras zawiera mutację punktową hamującą aktywność GTP-azy. Zmutowane onkogenne Ras nie są zatem zdolne do hydrolizy GTP nawet w obecności GAP i wskutek tego pozostają trwale pobudzone. Wysyłany przez nie sygnał mitogenny nie jest zależny on obecności właściwego liganda w środowisku komórki.
Mutacje genu ras występują w wielu nowotworach, w tym w ponad połowie wszystkich przypadków raka okrężnicy i 90% raków trzustki.
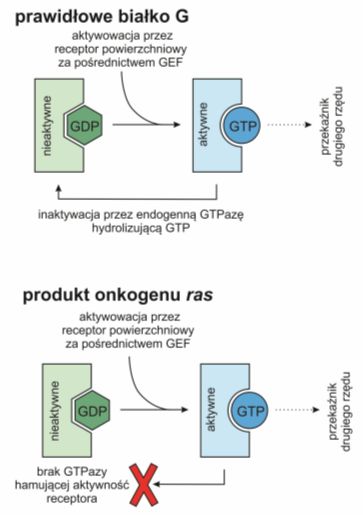
Rys.1 Kodowane przez onkogen ras białko nie posiada aktywnej GTP-azy i w związku z tym traci zdolność do samoregulacji.
• jądrowe czynniki transkrypcyjne, czyli białka regulujące ekspresję poszczególnych genów Swoje funkcje pełnią wiążąc się z DNA w obszarze promotora bądź sekwencji wzmacniającej
Gen c-myc jest protoonkogenem kodującym białko Myc, które wraz z białkiem Max tworzy heterodimer pełniący rolę czynnika transkrypcyjnego genów kontrolujących wzrost i różnicowanie komórek, angiogenezę i apoptozę.
Gen c-myc jest zlokalizowany na chromosomie 8 w pobliżu miejsca łamliwego. Jego nadmierna aktywacja zachodzi w wyniku pęknięcia w pozycji 8q24 i translokacji fragmentu chromosomu 8 na chromosom 2,14 lub 22. Mutacja ta prowadzi do 2-5 krotnego wzrostu ilości mRNA kodującego białko Myc. Zwiększona ilość białka powoduje powstawanie nowotworu.
Tego typu rearanżacje zachodzą m.in. w chłoniaku Burkitta, gdzie gen c-myc przenoszony jest na chromosom 14, w pobliże genu kodującego ciężki łańcuch immunoglobulin.
Aktywacja protoonkogenu c-myc może zachodzić również w wyniku amplifikacji genu. Występuje ona m.in. w raku trzustki, żołądka, okrężnicy, drobnokomórkowym raku płuca oraz raku sutka. Odgrywa przy tym ważną role w progresji nowotworu, gdyż wiąże się z jego zwiększoną agresywnością i zdolnością do metastazy.
Onkogeny jun i fos – kodują podjednostki prawidłowych czynników transkrypcyjnych Jun i Fos tworzących czynnik AP-1. W prawidłowych komórkach ekspresja obu genów zachodzi zaraz po stymulacji mitogennej i trwa bardzo krótko. Stężenie w komórce produktów Jun i Fos jest kontrolowane nie tylko na poziomie transkrypcji genu, ale również poprzez procesy wpływające na stabilność mRNA. W komórkach nowotworowych działanie obu tych mechanizmów zostaje zwielokrotnione.
Uzależnienie od onkogenów
Niektóre typy raka występują we wczesnym etapie życia i są związane z mutacją zachodzącą w pojedynczym genie. Przykładem może być siatkówczak (retinoblastoma), którego przyczyną jest mutacja genu supresorowego nowotworu – RB1. W większości przypadków proces transformacji nowotworowej jest jednak o wiele bardziej skomplikowany i składa się z licznych, postępujących po sobie etapów. Oznacza to, że pojedynczy onkogen nie jest w stanie wywołać pełnego zakresu zmian potrzebnych do przekształcenia się prawidłowej komórki w złośliwą, tzn. nowotworową.
Onkogeneza obejmuje szereg mutacji obniżających aktywność genów supresorowych i zwiększających aktywność protoonkogenów. Dodatkowo, komórki rakowe prawie zawsze cechują się zmienioną liczbą chromosomów, która wpływa na ilość i aktywność zlokalizowanych w nich genów.
Ilość zmian zachodzących w komórkach nowotworowych jest na tyle duża, że wręcz niemożliwy wydawał się pomysł, aby można je było wykorzystać jako molekularne cele terapii antyrakowej. Obecnie jednak wiadomo, że wzrost i proliferacja niektórych nowotworów złośliwych jest determinowana ekspresją konkretnych genów i zależy od nich w większym stopniu niż od pozostałych zmian. Jest to tzw. hipoteza uzależnienia od onkogenów (ang. oncogene addiction). Zablokowanie owego kluczowego genu zahamowałoby wzrost komórek i ograniczyło rozwój guza, a także, w niektórych przypadkach, zwiększyło wrażliwość na chemioterapeutyki. Poznanie onkogenów odgrywających kluczową rolę w formowaniu się danego typu nowotworu jest zatem istotne dla skutecznej walki z chorobą.
Izolacja onkogenów
Izolację onkogenów przeprowadza się za pomocą transfekcji obcym DNA komórek z linii NIH-3T3, czyli ustalonej linii komórkowej wyprowadzonej z fibroblastów mysich. Są to komórki tkanki łącznej, które bardzo dobrze pobierają i poddają ekspresji obcy materiał genetyczny. W warunkach in vitro rosną w taki sam sposób, co komórki nienowotworowe, to znaczy ulegają podziałom aż do momentu, gdy pokryją cała powierzchnię naczynia. Jest to tak zwana inhibicja kontaktowa.
Po dokonaniu transfekcji, komórki posiadające obcy DNA z wbudowanym onkogenem charakteryzują się wzrostem typowym dla komórek nowotworowych i kontynuują podziały formując w ten sposób wielowarstwowe skupiska.
Metoda ta jest o wiele prostsza i szybsza niż testy wykonywane w warunkach in vivo. Wykorzystanie linii komórkowych, nie zaś całego zwierzęcia, jest również bardzo przydatne podczas przeprowadzania testów przesiewowych dużej liczby próbek.
Technika ta nie jest jednak pozbawiona wad. Między innymi, ze względu na zastosowanie fibroblastów nie pozwala ona na wykrywanie niektórych onkogenów, charakterystycznych dla innych typów komórek. Przykładowo komórki NIH-3T3 mogą zostać pominięte przez geny zaangażowane we wczesne stadia kancerogenezy. Metoda nie pozwala również na wykrywanie genów supresji transformacji nowotworowej. Ponadto duże geny mogą nie zostać przeniesione lub zostać naruszone podczas dokonywania transfekcji.
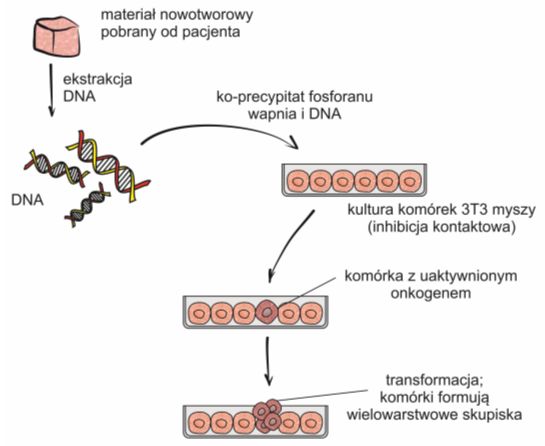
Rys. 2. Zmiana we wzroście komórek NIH-3T3 stanowi dowód na obecność onkogenu w ich DNA.
Autor: Anna Kurcek
Literatura:
1. Chial H., 2008. Proto-oncogenes to Oncogenes to Cancer. Nature Education, 1(1).
2. Chrzan P., Kowara R., Skokowski J., Gołębiowski F., Karmoliński A., Roszkiewicz A., Kopacz A., Pawełczyk T., 1999. Amplifikacja onkogenu c-myc i aberracje transkryptu genu FHIT w raku sutka. Onkol. Pol., 2 (4): 191-195.
3. Genetyczne podstawy chrobry nowotworowej; http://leczenie-objawy.pl/
4. Goodsell D. S., 1999. The Molecular Perspective: The ras Oncogene. The Oncologist, 4(3): 263-264.
5. Mantur M., Koper O., 2008. Płytkopochodny czynnik wzrostu – budowa, rola i jego receptory. Por. Merk. Lek., 24(140): 173-176.
6. Tokajuk P., Czartoryska-Arłukowicz B., Wojtukiewicz M. Z., 2011, Leczenie interferujące z funkcją EGFR u chorych na raka piersi. Onkologia w Praktyce Klinicznej, tom 7, nr. 4: 159–176.
7. Turner P.C., McLennan A.G., Bates A.D., White M.R.H., 2004. Krótkie wykłady – Biologia Molekularna. Wydawnictwo Naukowe PWN.
8. Winter P.C., Hickey G.I., Fletcher H.L., 2004. Krótkie wykłady – Genetyka. Wydawnictwo Naukowe PWN.