
Autor: Dr Tomasz Lech
Katedra Mikrobiologii
Uniwersytet Ekonomiczny w Krakowie
Mikroorganizmy to organizmy wszędobylskie zasiedlające wszystkie środowiska, w których znajdą szansę na przeżycie. Występują one w miejscach bogatych w związki organiczne, stanowiące dla nich źródło pożywienia, takich jak gleba, wody powierzchniowe czy też organizmy wyższe, jak również w miejscach skrajnie niesprzyjających dla organizmów żywych, między innymi w gorących źródłach, wodach o wysokim stężeniu soli, czy też głęboko w skorupie ziemskiej. Zarówno bakterie jak i grzyby charakteryzuje zdolność do wzrostu w szerokim spektrum temperatur (począwszy od temperatur ujemnych do temperatur nawet powyżej 100oC), wilgotności oraz stężenia jonów wodorowych w środowisku – pH). Największym problemem współczesnych mikrobiologów jest ciągle niski procent mikroorganizmów, które udaje się wyhodować w warunkach laboratoryjnych. Szacuje się, że zaledwie 5% mikroorganizmów środowiskowych potrafimy wyhodować na podłożach mikrobiologicznych. Jest to istotne ograniczenie zwłaszcza w badaniach bioróżnorodności mikrobiologicznej różnych środowisk, jak i identyfikacji poszczególnych taksonów bakteryjnych i grzybowych. Na przeciwko tym problemom wychodzi biologia molekularna z szerokim wachlarzem zaawansowanych technik badawczych, do których należą głównie metody oparte na łańcuchowej reakcji polimerazy (ang. polimerase chain reaction, PCR).
Pewną modyfikacją techniki PCR jest metoda PCR-DGGE (ang. polymerase chain reaction – denaturing gradient gel electrophoresis) – czyli reakcja łańcuchowa polimerazy w połączeniu z elektroforezą w gradiencie czynnika denaturującego. Umożliwia ona pozyskanie informacji o strukturze genotypowej mikroorganizmów obecnych w badanym środowisku na podstawie analizy różnic w wybranych sekwencjach genomu. Największą zaletą tej metody jest nie tylko możliwość badania różnorodności mikroorganizmów, ale także śledzenia jej zmian w czasie, a także możliwość identyfikacji gatunkowej poszczególnych taksonów bez konieczności ich hodowli.
PCR-DGGE
I etap
Pierwszym etapem badań jest izolacja materiału genetycznego z próbki pobranej ze środowiska. Materiał do badań może stanowić próbka gleby, wody, a nawet próbka powietrza, które jest medium przenoszenia mikroorganizmów w środowisku. Do izolacji DNA z mikroorganizmów, przez długi czas stosowano wieloetapową metodę opartą na trawieniu ich komórek w buforze lizującym z dodatkiem enzymów degradujących ich ściany i inne organelle komórkowe. Otrzymany w ten sposób lizat poddawano oczyszczaniu w układzie fenol – chloroform, a DNA wytrącano alkoholem izopropylowym. Obecnie do izolacji kwasów nukleinowych najczęściej stosuje się komercyjnie dostępne zestawy kolumienkowe, oparte na wykorzystaniu membran celulozowo-krzemionkowych selektywnie wiążących DNA. W przypadku tej metody również wykorzystuje się degradację komórek w buforze lizującym z dodatkiem enzymów trawiących, następnie otrzymany lizat przenosi się na kolumnę z membraną, która w trakcie wirowania wiąże DNA. Po kilku krotnym przemyciu, buforami płuczącymi, materiału genetycznego związanego do membrany prowadzona jest jego elucja. Należy zwrócić uwagę, iż w przypadku próbek do PCR-DGGE izoluje się DNA stanowiący mieszaninę materiału genetycznego wszystkich obecnych organizmów w próbce.
II etap
Kolejnym etapem w metodzie PCR-DGGE jest amplifikacja wybranych sekwencji genomowych mikroorganizmów, na podstawie których dokonywane będzie ich różnicowanie. Jak już wspomniano wcześniej w próbce przygotowanego materiału genetycznego jest mieszanina DNA wielu mikroorganizmów, dlatego też aby uzyskać wyniki najpełniej opisujące badaną mikroflorę, dobór odpowiednich sekwencji jest bardzo ważny. W przypadku bakterii najczęściej wykorzystuje się sekwencje w obrębie genu 16S rDNA, natomiast w przypadku grzybów są to sekwencje w niekodującym regionie ITS (ang. Internal Transcribed Spacer). Łańcuchową reakcję polimerazy prowadzi się w standardowych warunkach zarówno dla mieszaniny reakcyjnej jak i programu z wykorzystaniem aparatu termocyklera umożliwiającego płynną zmianę temperatury (Fot.1).
Mieszanina PCR: H2O, 10x stężony bufor do polimerazy, MgCl2, startery DNA, mieszanina oligonukleotydów, polimeraza Taq oraz matryca DNA.
Warunki temperaturowe prowadzenia PCR: denaturacja wstępna – zazwyczaj 95oC przez 5-10 min, denaturacja – 95oC przez 30-60 s, przyłączanie starterów DNA do matrycy – temperatura zależna od stosowanych starterów DNA najczęściej przez 30-100 s, elongacja/synteza łańcucha DNA – 72oC najczęściej przez 30-120 s, ostateczna elongacja – 72oC.
Dla próbek o małej zawartości DNA, jak to często jest w przypadku próbek powietrza, możliwe jest zastosowanie techniki nestedPCR (PCR zagnieżdżony). W metodzie tej amplifikację wybranych sekwencji materiału genetycznego przeprowadza się w dwóch reakcjach polimerazy następujących po sobie. W pierwszym PCR matrycę stanowi DNA wyizolowane z próbki środowiskowej, natomiast w drugim PCR matrycą są produkty otrzymane w pierwszym PCR.

Fotografia 1. Termocykler firmy Bio-Rad umożliwiający amplifikację materiału genetycznego. Fot. Tomasz Lech
III etap
Po uzyskaniu odpowiedniej jakości amplikonów przeprowadza się ich elektroforezę w żelu poliakrylamidowym (akrylamid:bisakrylamid, 39.5:1) z gradientem czynnika denaturującego najczęściej 30-60% (możliwe 20-80%), którym najczęściej jest mocznik. Bufor do elektroforezy stanowi 0,5% bufor TAE. Po przeprowadzonej elektroforezie żel wybarwia się (bromkiem etydyny lub komercyjnie dostepnymi barwnikami), a otrzymany wzór prążków (fingerprint) wizualizuje się z wykorzystaniem światła UV. Napięcie i czas prowadzenia elektroforezy zależny jest od wielkości rozdzielanych amplikonów (Fot. 2).
Bufor TAE (na 1 litr):
– Tris – 242 g
– Kwas octowy lodowaty – 57,1 ml
– 0.5 M EDTA (pH 8.0) – 100 ml
Żel poliakrylamidowy 8% o 100% stężeniu czynnika denaturującego (100 ml):
– 40% akrylamid/bisakrylamid – 20 ml
– Mocznik – 42 g
– Formamid (100%) – 40 ml
– 50x TAE 2 ml
– Woda filtrowana do 100 ml
W celu uzyskania żelu o pożądanym stężeniu czynnika denaturującego należy proporcjonalnie zmieniać zawartość mocznika i formamidu, a w przypadku żelu o 0% zawartości czynnika denaturującego składniki te należy zupełnie pominąć. Do polimeryzacji żelu wykorzystuje się 10% roztwór APS (Ammoniumpersulfate) oraz TEMED (Tetramethylethylenediamine).

Fotografia 2. Zestaw do DGGE firmy Bio-Rad. Fot. Tomasz Lech
Przykładowy PCR-DGGE fingerprint przedstawiono na fotografii 3. Fotografia przedstawia wzory prążków uzyskane w badaniach różnorodności genotypów grzybowych obecnych w powietrzu czterech różnych środowisk. Do uzyskania amplikonów PCR wykorzystano technikę nestedPCR, gdzie w pierwszej reakcji wykorzystano startery ITS1F i ITS4, natomiast druga reakcja przebiegała z wykorzystaniem starterów ITS1F-GC i ITS2. Charakterystyczne dla metody PCR-DGGE jest stosowanie starterów DNA z ogonem GC (odcinek o długości ok 40 nukleotydów cytozynowych i guaninowych), zapobiega on całkowitej denaturacji rozdzielanych amplikonów podczas elektroforezy.
Po uzyskaniu fingerprintu możliwa jest identyfikacja gatunkowa poszczególnych taksonów. W tym celu wycina się pojedyncze prążki z żelu, przeprowadza się reamplifikację DNA z wykorzystaniem tych samych starterów lecz bez ogona GC, a uzyskany produkt poddaje się elektroforezie. Uzyskany prążek ponownie wycina się z żelu, oczyszcza przy wykorzystaniu komercyjnych zestawów do oczyszczania DNA z żelu i poddaje się sekwencjonowaniu. Ogólny przebieg badania z wykorzystaniem metody PCR-DGGE przedstawiono na Rysunku 1.
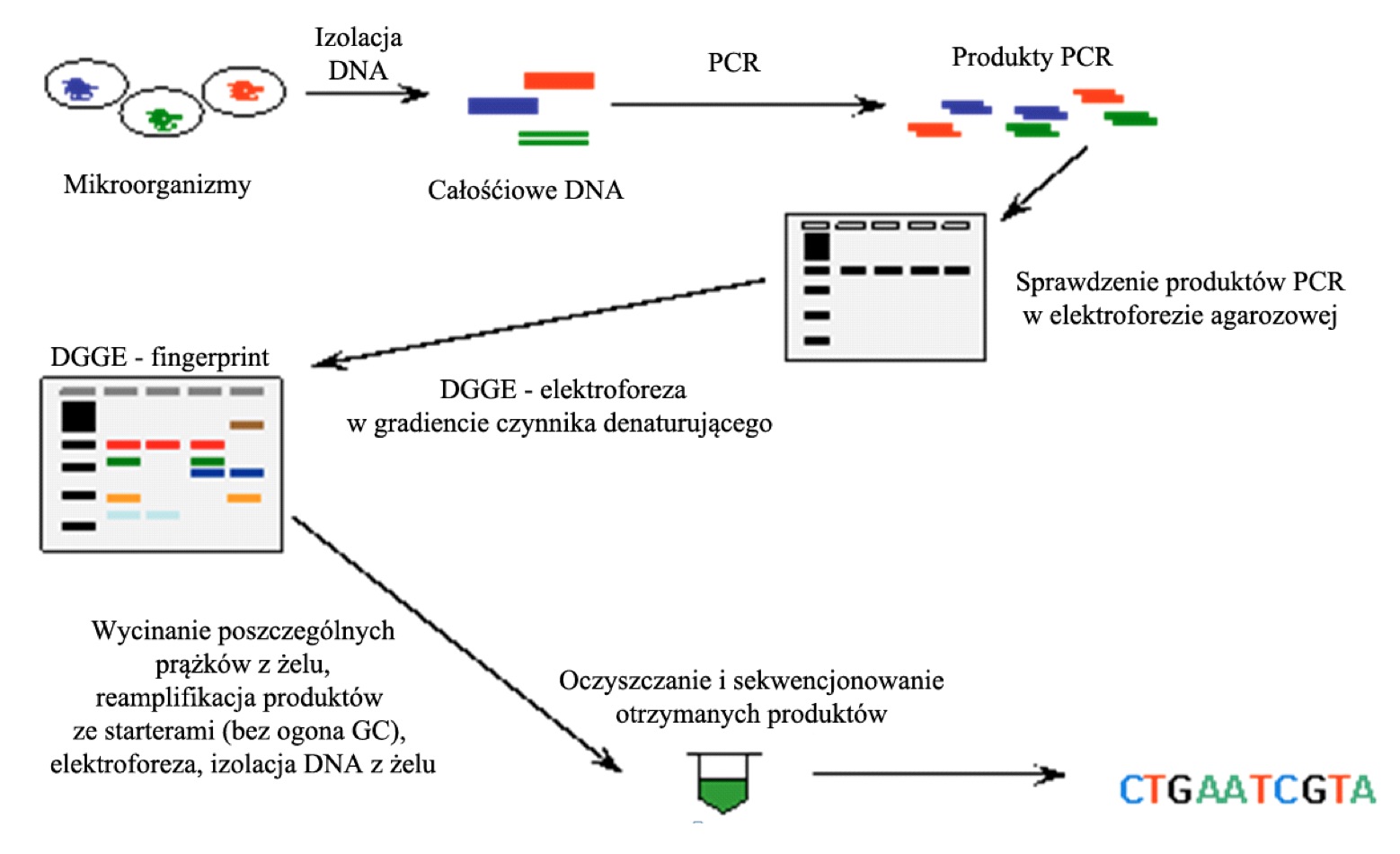
Rysunek 1. Schemat przedstawiający poszczególne etapy metody PCR-DGGE [Źródło: http://wiki.biomine.skelleftea.se/biomine/molecular/index_11.htm – dostęp na 02.04.2016 – ze zmianami]
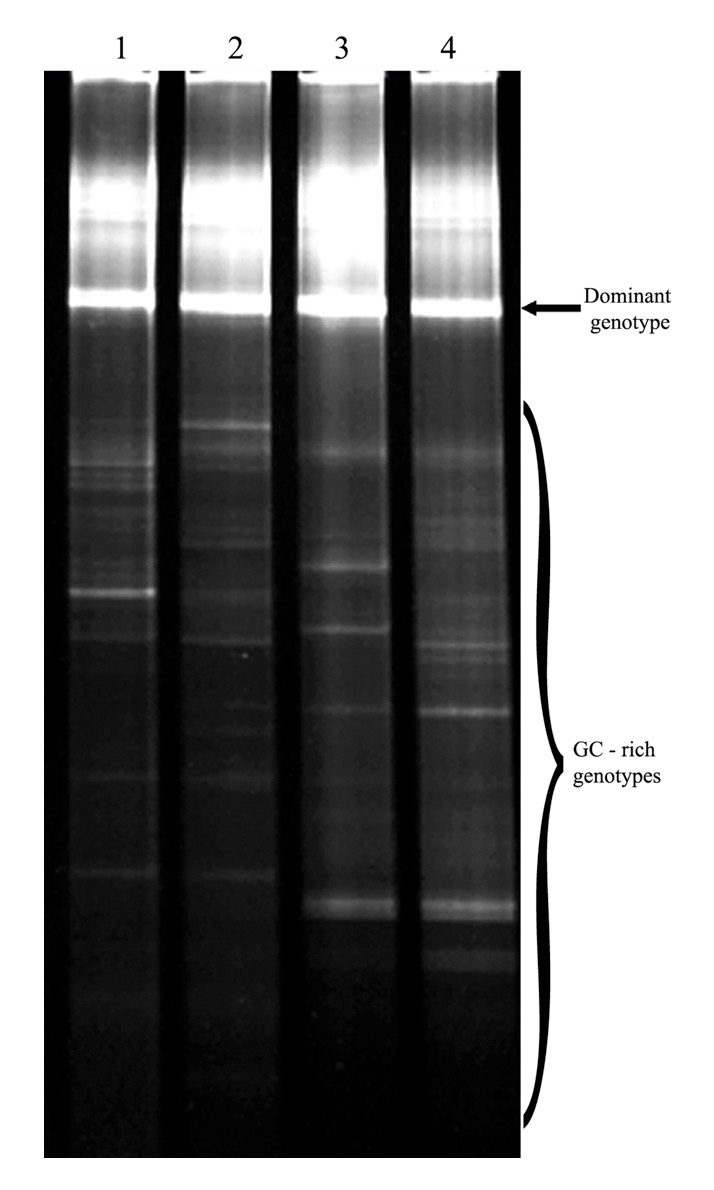
Fotografia 3. Przykładowy PCR-DGGE fingerprint. [Lech i in., 2014]
Literatura:
1. Błaszczyk M.K., 2010, „Mikrobiologia środowisk”. Wydawnictwo Naukowe PWN, ss. 400.
2. Muyzer G, de Waal EC, Uitterlinden AG. Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Appl Environ Microbiol. 1993;59(3):695-700.
3. Muyzer G, Smalla K. Application of denaturing gradient gel electrophoresis (DGGE) and temperature gradient gel electrophoresis (TGGE) in microbial ecology. Antonie van Leeuwenhoek. 1998;73(1):127-141.
4. Lech T, Ziembinska-Buczynska A. Evaluation of a modified sampling method for molecular microbial communities. Genet Mol Res. 2015;14(2):3200-3208.
5. Ziembińska-Buczyńska A, Wiszniowski J, Ciesielski S. Comparison of PCR-DGGE and Nested-PCR-DGGE Approach for Ammonia Oxidizers Monitoring in Membrane Bioreactors’ Activated Sludge. Arch Env Prot. 2014;40(4):31-38.
6. http://wiki.biomine.skelleftea.se/biomine/molecular/index_11.htm – dostęp na 02.04.2016 – ze zmianami