
Termin „priony” (ang. proteinaceous infectious particle) został po raz pierwszy użyty w XX w. jako określenie białkowych cząsteczek zakaźnych, które mimo braku metabolizmu oraz kwasów nukleinowych, zdolne są do samopowielania się w organizmie gospodarza. U ssaków mogą one wywoływać przewlekłe śmiertelne zwyrodnienia tkanki nerwowej mózgu, czyli tzw. encefalopatie gąbczaste. Odkrycie grzybowych prionów i ich podobieństwo do ssaczych odpowiedników pozwala jednak przypuszczać, że te samoreplikujące się białka nie są biologiczną anomalią, lecz jednym z mechanizmów regulujących fenotyp komórkowy.
Początki odkryć
Choroby prionowe były powszechnie znane już w XVIII w., jednak natura wywołujących je czynników infekcyjnych pozostawała niewyjaśniona aż do wieku XX.. Początkowo, ze względu na odporność na promieniowanie jonizujące i ultrafioletowe (UV), schorzeniom tym przypisywano pochodzenie wirusowe. Dopiero w 1967 r. John Stanley Griffith jako pierwszy wysunął przypuszczenie, że wywołujący je patogen może nie posiadać własnego materiału genetycznego i być jedynie zmienioną formą któregoś z białek komórkowych. Przypuszczenia te potwierdzono na początku lat 80- tych, kiedy to Stanley Prusiner wyizolował z mózgu zakażonego scrapie zwierzęcia czynnik chorobotwórczy i opisał jego właściwości: odporność na podgrzewanie, traktowanie proteinazą K, mocznikiem, solami chalotropowymi, SDS (ang. sodium dodecyl sulfate) oraz takimi czynnikami niszczących DNA, jak nukleazy i psoralen. Odkryto również, że w obecności tlenu pozostaje on niewrażliwy na działanie promieniowania jonizującego, a zatem wykazuje właściwość typową dla białek hydrofobowych związanych z lipidami. Tak scharakteryzowany nowy czynnik infekcyjny nazwano „prionem” (ang. proteinaceous infectious particle), czyli białkową cząstką zakaźną.
PrP
Czynnikiem zakaźnym wywołującym scrapie okazało się być białko błonowe PrP (ang. Prion Protein), występujące głównie w komórkach centralnego układu nerwowego i tkance chłonno – siateczkowej. Kodujący je gen Prnp jest obecny u ssaków, ptaków oraz ryb.
Fizjologiczną postać PrP oznacza się jako PrPC, formę patologiczną, tj. odpowiedzialną za występowanie infekcji, jako PrPSc (od „scrapie”). I choć obie postacie białka mają identyczną sekwencję aminokwasową, to różnią się między sobą strukturą przestrzenną. Dzięki badaniom z wykorzystaniem rezonansu magnetycznego wykazano, że cząsteczka PrPC zawiera 42% α-helis i jedynie 3% β- struktur, podczas gdy PrPSc składa się z 30% α-helis i 43% β struktur. Pozwala to przypuszczać, że właściwości infekcyjne prionów wynikają z rearanżacji ich struktury.
Teoria prionowa
Bazując na danych eksperymentalnych dostępnych w 1982 r. Stanley Prusiner sformułował tzw. teorię prionową, według której:
– poszukiwanym czynnikiem infekcyjnym jest białko PrPSc;
– PrPSc może się samodzielnie namnażać, mimo braku kwasów nukleinowych;
– przejście fizjologicznej postaci białka PrP (PrPC) w formę infekcyjną (PrPSc) polega na zmianie konformacji;
– choroby degeneracyjne tkanki nerwowej mogą być powodowane spontaniczną zmianą konformacji z PrPC do PrPSc (sporadyczna, samoistna choroba prionowa), przedostaniem się do organizmu form patologicznych pochodzących z zewnątrz (nabyta, przepasażowana choroba prionowa) lub mutacją genu Prnp (dziedziczna, rodzinna choroba prionowa).
Zgodnie za założeniami tej teorii organizm pozbawiony PrPC powinien być odporny na infekcję prionami. Słuszność tego założenia potwierdzono przeprowadzając badania na transgenicznych myszach będących homozygotami dla delecji Prnp (Prnp 0/0). Wprowadzenie do ich organizmów homogenatu mózgowego pochodzącego ze zwierząt zakażonych scrapie nie przyczyniło się do replikacji prionów, ani też do zniszczenia tkanki nerwowej. W ten sposób wykazano również, że PrPC odpowiada za dostarczenie patogenu z nerwów peryferyjnych do centralnego systemu nerwowego.
Teoria prionowa została potwierdzona na drodze eksperymentalnej i przyjęta przez większość naukowców.
Materiał do badań
Do dalszych analiz nad infekcyjnością prionów potrzebne było opracowanie sposobu umożliwiającego otrzymywanie niezbędnej ilości materiału badawczego. Problem ten rozwiązano dzięki systemowi cyklicznej amplifikcji, w którym jako oryginalną matrycę wykorzystuje się patologiczne białko PrPSc izolowane z mózgowego homogenatu zainfekowanego zwierzęcia. Homogenat rozcieńcza się 104 razy i inkubuje z nadmiarem białka PrPC. W ten sposób uzyskuje się agregaty formy infekcyjnej, które następnie rozbija się za pomocą sonikacji, ponownie rozcieńcza i inkubuje z PrPC. Proces ten powtarza się, aż do momentu uzyskania roztworu o stężeniu 1055. W kolejnych cyklach matrycami są powstałe in vitro formy infekcyjne, które pod względem budowy biochemicznej nie różnią się od PrPSc wyizolowanego z mózgu zwierzęcia. Również ich doczaszkowa iniekcja powoduje występowanie identycznych zwyrodnień gąbczastych tkanki mózgowej. Jednak z niewyjaśnionych przyczyn, białko otrzymywane in vitro jest mniej infekcyjne niż PrPSc uzyskiwane in vivo.
Obecnie białko PrPC produkuje się z udziałem bakterii (ang. recombinant prion protein – rPrP). Forma ta może być otrzymywana w dużych ilościach i pomimo pewnych różnic w budowie (nieobecności glikanów i kotwicy GPI) posiada taką samą strukturę drugo- i trzeciorzędową, co PrPC wyizolowany z mózgu. Dzięki temu stanowi ona użyteczne narzędzie stosowane do badań nad fizykochemicznymi właściwościami i zmianami konformacyjnymi białek prionowych.
Mechanizm konwersji prionów
Istnieje kilka modeli wyjaśniających mechanizm przekształcania PrPC do PrPSc.
Pierwszym z nich to model heterodimeru (ang. heterodimer model) lub inaczej heterodimeryczny mechanizm fałdowania, który zakłada, że formowanie się agregatów białkowych nie jest konieczne dla konwersji prionów. Ich tworzenie się jest rozpatrywane jedynie jako proces wtórny, niezwiązany z rearanżację konformacyjną. Krytycznym punktem przemiany formy fizjologicznej w infekcyjną jest formowanie się heterodimeru złożonego z monomerów PrPC i PrPSc. Forma infekcyjna pełni rolę monomerycznej matrycy indukującej zmianę konformacji PrPC.
Odwrotne założenia wynikają z modelu polimeryzacji (ang. polimerization model), według którego agregacja jest niezbędna do stabilizacji formy PrPSc. Przemiana prionów i polimeryzacja są zatem procesami nierozerwalnymi. Etapem limitującym przejście PrPC w PrPSc jest formowanie się oligomerów PrPSc, czyli tzw. „ziaren” (ang. “seed”) stanowiących produkty pośrednie przemiany prionowej. Zaproponowano dwa rodzaje tego modelu. Wariant pierwszy zakłada, że zmiany konformacyjne monomerów PrPC i PrPSc mają miejsce jeszcze przed ich przyłączeniem się do oligomeru. Związanie z oligomerem pełni funkcje stabilizacyjne, a monomer PrPSc, który nie zwiąże się z ziarnem przemienia się w PrPC. Według drugiego schematu zmiany w strukturze monomerów zachodzą dopiero w momencie ich przyłączenia się do oligomeru PrPSc.
Już po sformułowaniu tych teorii okazało się, że półprodukty oligomeryczne przypominają budową micele, a zatem mają strukturę mniej zorganizowaną niż typowe włókna amyloidowe. Odkrycie to przyczyniło się do wzbogacenia drugiego wariantu modelu polimeryzacyjnego o dodatkowe założenia. Ustalono, że jedynie stabilne ziarna mogą służyć jako matryce do budowy prionów. Wiążą się z nimi nie tylko monomery, ale także kompleksy oligomerów, które w ten sposób nabywają nową strukturę konformacyjną i zyskują zdolność do katalizowania przemian prionowych.
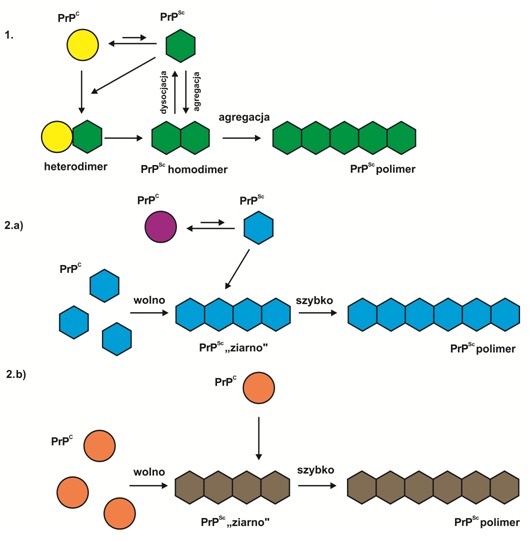
Rys.1 Schematy obrazujące różne mechanizmy tworzenia się prionów: model heterodimeryczny (1) oraz wariant pierwszy (2. a) i drugi (2. b) modelu polimeryzacyjnego; opis w tekście
Choroby prionowe
Choroby prionowe, czyli gąbczaste encefalopatie (ang. transmissible spongiform encephalopathies – TSE) dotykają średnio jednego człowieka na milion i mogą być dziedziczone (ok. 15%), nabyte (<1%) lub powstawać spontanicznie (85%). Jak dotąd uznawane są one za śmiertelne i niezależnie od pochodzenia wykazują właściwości zakaźne.
Infekcyjność prionów została po raz pierwszy udowodniona przez R. Chandlera, który zaraził mysz chorobą typową dla owiec – scrapie, a następnie przez D. C. Gajduseka, który zainfekował szympansa ludzką encefalopatią – kuru. Do zakażenia ssaczymi prionami dochodzi zwykle w wyniku spożycia mięsa, głównie mózgu chorego osobnika. Przeniesienie czynników infekcyjnych może nastąpić również poprzez niedokładną sterylizację narzędzi chirurgicznych. Zakażenia eksperymentalne przeprowadza się na drodze dootrzewnowej lub wewnątrzczaszkowej iniekcji homogenatu sporządzonego z mózgu chorego zwierzęcia.
Tab.1 Choroby prionowe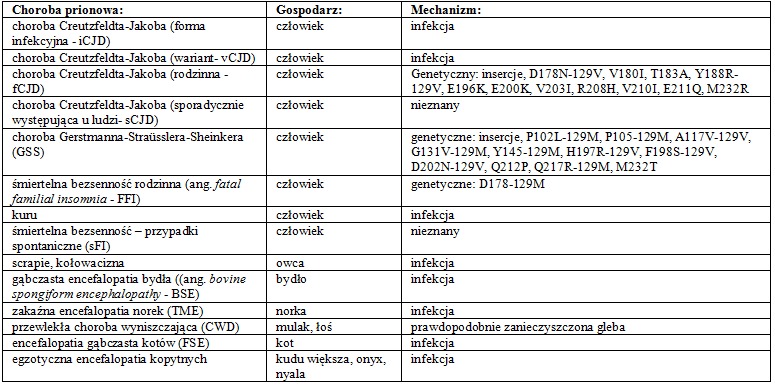
Objawy kliniczne gąbczastych encefalopatii ujawniają się po dość długim okresie inkubacji i obejmują różnego rodzaju zaburzenia ruchowe oraz poznawcze. W mózgach zakażonych osobników obserwuje się charakterystyczne gąbczaste degeneracje, astrogliozę, a także nagromadzenie nie natywnych złogów białkowych, tzw. włókien amyloidowych. Różnorodność objawów oraz czas inkubacji w dużym stopniu zależą od szczepu prionu.
Dokładna natura neurotoksyczności prionów pozostaje niejasna. Nie widomo, czy jest ona powodowana utratą funkcji PrPC, czy raczej toksycznymi właściwościami tworzącego się PrPSc. Możliwe również, że w grę wchodzą zupełnie inne czynniki, kierujące neurony na jedną z dróg programowanej śmierci komórkowej spowodowanej stresem oksydacyjnym, regulowaną aktywacją dopełniacza, szlakami ubikwitynowo-proteosomowym lub endosomalno – lizosomowym, zmianami synaptycznymi, atrofią dendrytyczną, odpowiedzią kortykosteroidów bądź też stresem retikulum endoplazmatycznego.
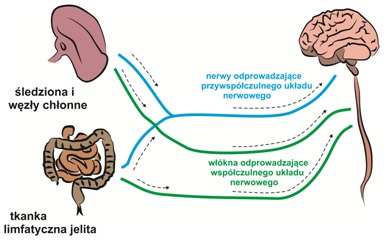
Rys.2 Schemat przedstawiający prawdopodobny przebieg inwazji układu nerwowego przez priony
Włókna amyloidowe
Choroby prionowe związane są z odkładaniem się tzw. włókien amyloidowych. Tego typu twory obserwuje się również w przypadku innych schorzeń neurodegeneracyjnych, takich jak choroba Alzheimera, Huntingtona i Parkinsona. W każdym powyższym przypadku, fibrylarne agregaty są zbudowane z białek, które w normalnych warunkach ulegają rozpuszczeniu. Ich odkładanie się i formowanie złogów są spowodowane zmianami w strukturze przestrzennej. Zwyrodnienia tego typu określa się mianem schorzeń amyloidowych lub inaczej konformacyjnych.
Zdolność do tworzenia amyloidów zależy od ich ładunku oraz hydrofobowości białek. U samego człowieka odkryto około 20 różnych cząsteczek zdolnych do formowania tego typu struktur w warunkach in vivo, m.in. insulinę, fragmenty lekkich łańcuchów przeciwciał, α-synukleinę, β-peptyd, a także białko PrP.
Wszystkie amyloidy wykazują niewrażliwość na trawienie proteolityczne, nierozpuszczalność i powinowactwo do aromatycznych barwników takich jak czerwień Kongo, czy tioflawina T. Rodzaj budującego je białka nie ma znaczenia.
Twory te są do siebie podobne również pod względem strukturalnym. Pojedyncze włókno stanowi pustą nanorurkę o średnicy 3nm i zawiera kilka splecionych ze sobą i ułożonych spiralnie protofibryli. Na jeden skręt takiej helisy przypada dwadzieścia reszt aminokwasowych. Aby utworzyć trwałą strukturę potrzebne są minimum dwa obroty, tzn. fragment amyloidu musi zawierać co najmniej czterdzieści reszt aminokwasowych. Całość stabilizują wiązania wodorowe tworzące się pomiędzy łańcuchem głównym i łańcuchami bocznymi (każdy -CO poprzedniego skrętu łączy się z -NH skrętu następnego). Orientacja łańcuchów bocznych aminokwasów zmienia się w każdym obrocie, w taki sposób, że sąsiadujące ze sobą łańcuchy boczne są ulokowane po różnych stronach łańcucha głównego, tzn. na zewnątrz lub do środka helisy.
Sekwencja aminokwasowa prionów
Wszystkie amyloidogenne sekwencje białkowe cechują się dużą zawartością reszt asparaginowych (Asn) i glutaminowych (Gln). Odkrycie to sugeruje możliwość przewidywania właściwości infekcyjnych białek na podstawie ich struktury pierwszorzędowej. Jako kryterium identyfikacji prionów stosuje się obecność 30-u reszt glutaminowych lub asparaginowych występujących w ciągłej sekwencji 80-u reszt aminokwasowych (ang. prion-forming domain -PrD). Trzeba jednak pamiętać, że nie wszystkie sekwencje formujące priony są bogate w tego typu układy i z tego powodu kryterium to nie jest narzędziem niezawodnym, np. nie dotyczy ono domen białek PrP i HET (Podospora anserina).
Priony u niższych eukariontów
Pierwsze priony grzybowe [Het-s] odkryto u Podospora anserina. W odróżnieniu od swoich ssaczych odpowiedników nie powodują one śmierci komórki gospodarza, lecz pozwalają zwiększyć jej zdolności dostosowawcze i umożliwiają przeżycie w niekorzystnych warunkach środowiskowych. Wiele białek prionowych odgrywa znaczącą rolę w procesie regulacji genów na poziomie transkrypcyjnym i potranskrypcyjnym i w ten sposób pośrednio wpływa na procesy komórkowe.
W 1994 r. R. Wicker wykorzystał teorię prionową do wyjaśnienia natury dwóch czynników występujących u drożdży z gatunku Saccharomyces serevisiae: [URE3] i [PSI+]. Priony drożdżowe nazwał białkami działającymi jak geny, podkreślając w ten sposób ich zdolność do gromadzenia i przekazywania konformacyjnej informacji. Po genetycznym skrzyżowaniu pomiędzy haploidalnymi szczepami [PRION+] i [prion-] Saccharomyces cerevisiae, typowym powstającym diploidem był osobnik [PRION+]. Przekazywał on czynnik prionowy całemu swojemu potomstwu. Jednakże, jeżeli związana z obecnością prionu cecha była kontrolowana przez mutację powodującą inaktywację jakiegoś genu jądrowego, wtedy prionowy fenotyp występował w diploidzie jako cecha recesywna, a wśród uzyskanego potomstwa widoczny był rozkład 2[PRION+]: 2[prion-].
W większości przypadków dziedziczenie czynnika prionowego nie jest związane ze zmianami w sekwencji nukleotydowej i prowadzi do ustanowienia się nowego, stabilnego stanu, który może się utrzymywać w populacji przez wiele pokoleń.
Stanowi to dowód na to, że priony są dziedziczone na zasadzie transferu przez cytoplazmę, czyli w ten sam sposób co małe mutacje mitochondrialne. Można je zatem uznać za czynniki epigenetyczne, które dzięki szybkim zmianom fenotypowym komórki, pozwalają na dostosowanie się do panujących warunków środowiskowych bez konieczności wprowadzania zmian w funkcjonowaniu genomu.
Tab.2 Priony u grzybów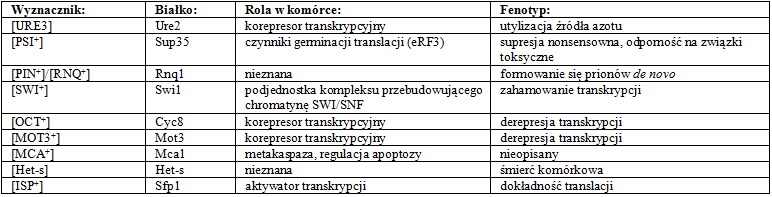
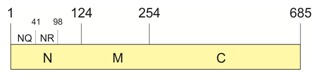
Rys. 3. Struktura białka Sup35, składającego się z regionów N i C przedzielonych regionem M. Domeny NR i NQ uznaje się za kluczowe dla właściwości prionowych białka
Szczepy prionów
Podstawową właściwością prionów jest tzw. zmienność szczepów. Ich ilość i układ wyznacza struktura pierwszorzędowa budującego je białka. Rodzaj konformacji wpływa na przebieg chorób prionowych, czas inkubacji, objawy kliniczne oraz obszary mózgu w których następują zmiany. Wszystkie szczepy zachowują swoje właściwości podczas namnażania się w zainfekowanych zwierzętach laboratoryjnych, to znaczy są stabilne w warunkach in vitro.
Istnienie różnych stanów konformacyjnych u prionów odkryto podczas przeprowadzania badań laboratoryjnych na chomikach zainfekowanych dwoma rodzajami gąbczastej encefalopatii norek: HY i DY. Różnią się one od siebie zarówno okresem wylęgania choroby, jak i objawami klinicznymi. Po częściowej proteolizie (z udziałem proteinazy K) PrPSc wyizolowanych z mózgów chomików zainfekowanych HY i DY, okazało się, że masa cząsteczkowa fragmentów HY odpornych na działanie proteaz była o 2kD większa niż analogiczny fragment w cząsteczce DY. Dalsze analizy wykazały różnice w drugorzędowej budowie ich β-struktury. Różnica między szczepami HY i DY polegała zatem na niejednakowej strukturze trójwymiarowej PrPSc.
Analizy różnych PrPSc wywołujących chorobę Creutzfelda-Jakoba wykazały, że szczepy prionów mogą różnić się między sobą również poziomem glikozylacji. PrP jest sialoglikoproteiną z dwoma miejscami N-glikozylacji w C-terminalnym regionie. Wyróżnia się zatem formy nie-, mono- i diglikozylowane. Wciąż nie wiadomo jednak, w jaki sposób zmiany te wpływają na budowę PrPSc i różnicują wywoływane objawy chorobowe.
Bariera międzygatunkowa
Szerzenie się infekcji prionowych jest ograniczane ze względu na występowanie tzw. barier międzygatunkowych. Gąbczaste encefalopatie przenoszone są jedynie pomiędzy osobnikami z tego samego lub blisko spokrewnionego gatunku. Przykładowo choroba Creutzfelda – Jakoba może przechodzić między ludźmi i szympansami. Te ostatnie nie mogą jednak zostać zakażone scrapie, czyli chorobą charakterystyczną dla owiec i kóz.
Bariery wewnątrzgatunkowe ujawniają się nie tylko jako niemożliwość przenoszenia choroby między niespokrewnionymi gatunkami, ale powodują także ujawnianie się choroby jedynie u części zainfekowanych osobników. Nie są one jednak absolutne i w niektórych przypadkach można je pokonać wydłużając czas inkubacji.
Przeniesienie choroby Creutzfelda-Jakoba z ludzi na myszy jest ograniczane przez barierę międzygatunkową, ale transgeniczne myszy produkujące ludzkie PrP są podatne na tego typu infekcje. Również transgeniczne myszy produkujące chomiczy PrP są bardziej wrażliwe na chorobę powodowaną przez chomicze priony, niż myszy typu dzikiego. Pozwala to przypuszczać, że obecność barier jest spowodowana różnicami w podstawowej strukturze PrP i może zostać zniesiona jedynie w przypadku, kiedy konformacje prionów u dwóch różnych gatunków (lub szczepów) nakładają się na siebie.
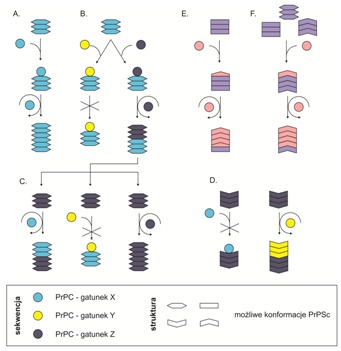
Rys. 4. Szczepy prionów i bariera międzygatunkowa. Poszczególne warianty sekwencji PrPC oznaczono różnymi kolorami, natomiast konformacje reprezentujące poszczególne szczepy prionów rozróżniono za pomocą kształtów. Zainfekowanie gatunku X odpowiednim prionem prowadzi do jego namnożenia i do powielania się cech danego szczepu (A.). Przepasażowanie namnożonego prionu na osobniki z gatunków Y i Z nieprodukujące homologicznych PrPC może prowadzić do wystąpienia bariery (jeżeli konformacja wprowadzonego PrPSc nie jest dostępna dla PrPC gospodarza, tak jak to ma miejsce w gatunku Y) lub infekcji, której wynikiem jest powstanie nowego szczepu prionów w zainfekowanym organizmie (jeżeli konformacja zaaplikowanego szczepu jest dostępna dla PrPC gospodarza – gatunek Z) (B.). Nowoutworzone szczepy prionów z gatunku Z, mogą przenosić cechy specyficzne dla pierwotnego wzorca (C.), lub tworzyć bariery inne niż te obserwowane w przypadku szczepu przeniesionego z gatunku X. Infekcyjność jest zatem związana z konformacją szczepu (D.). Podczas międzygatunkowych transmisji specyficznego szczepu prionu obserwuje się tzw. „przełączanie szczepu’ (ang. ”strain switching”): konwersję szczepu, kiedy to substrat PrPC dostosowuje swoją konformację do tej w niehomologicznej matrycy PrPSc (E.) lub selekcję szczepu, w której szczep chorobotwórczy związany z różnymi konformerami PrPSc, z których jeden jest dominujący w danym gatunku. Poprzez międzygatunkową transmisję, niehomologiczny PrPC gospodarza wybiera matrycę PrPSc najzgodniejszą z jego własną sekwencją aminokwasową. W każdym przypadku nowe konformacje PrPSc związane z pojawiającym się szczepem mogą powodować występowanie różnych barier i objawów chorobowych (F.)
Autor: Anna Kurcek
Literatura:
1. Tuite M. F., Serio T. R., 2010. The prion hypothesis: from biological anomaly to basic regulatory mechanism. Nat Rev Mol Cell Biol, 11(12): 823–833.
2. Shkundina I. S., Ter-Avanesyan M. D., 2007. Prions. Biochemistry (Moscow), 72 (13): 1519-1536.
3. Surewicz W. K., Cobb N. J., 2009. Prion Diseases and Their Biochemical Mechanisms. Biochemistry, 48(12): 2574–2585.
4. Hoope N. M., Kellett K. AB., 2009. Prion protein and Alzheimer disease. Prion, 3(4): 190–194.
5. Benetti F., Legname G., 2009. De novo mammalian prion synthesis. Prion, 3(4): 213–219.
6. Kovacs G. G., Budka H., 2008. Prion Diseases: From Protein to Cell Pathology. Am J Pathol, 172(3): 555–565.