
Od lat wiadomo, że sekwencja definiuje strukturę przestrzenną białka (hipoteza termodynamiczna, dogmat Anfinsena). Funkcja pełniona w komórce przez natywne białko jest zależna od sposobu zwinięcia łańcucha polipeptydowego i ułożenia grup bocznych aminokwasów.
Struktury przestrzenne białek otrzymywane metodami doświadczalnymi takimi, jak krystalografia rentgenowska czy spektroskopia NMR oraz struktury teoretyczne uzyskane w wyniku modelowania homologicznego czy de novo niosą ze sobą bardzo dużo informacji. Tego typu dane strukturalne mogą być wykorzystane przez biotechnologów i biologów molekularnych do uzyskania dodatkowych informacji i precyzyjnego projektowania doświadczeń.
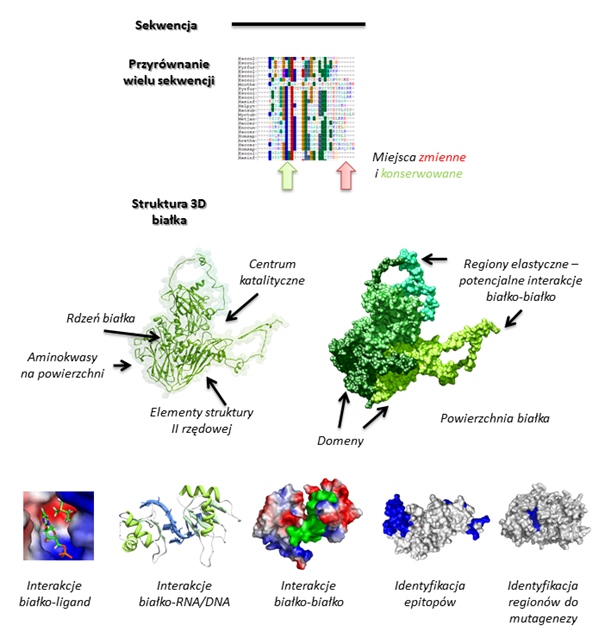
Co można zrobić posiadając dobrej jakości dane strukturalne? Oto kilka przykładów:
1. Identyfikacja reszt aminokwasów pod kątem projektowania eksperymentu mutagenezy. Znając jedynie sekwencję białka można dość losowo mutować aminokwasy i sprawdzać jaki efekt będziemy obserwować fenotypowo. Niestety, jest to proces żmudny i czasochłonny, gdyż wymaga wielu losowych prób. Aby ograniczyć losowość przewidywań i zdobyć więcej informacji można przygotować przyrównania posiadanej sekwencji do sekwencji białek homologicznych. Zidentyfikujemy dzięki temu regiony zmienne oraz konserwowane, a aminokwasy wybrane do mutagenezy mogą okazać się trafniejsze.
Natomiast, dysponując strukturą białka i danymi w niej zawartymi możemy badania skierować na nowy tor. Analiza oddziaływań białka z innymi białkami, bądź cząsteczkami DNA lub RNA jest możliwa dzięki identyfikacji reszt aminokwasowych, które znajdują się na powierzchni białka. Natomiast, na stabilność struktury można wpłynąć poprzez wprowadzenie mutacji w regionach sekwencji budujących elementy struktury II rzędowej (helisy-alfa i wstęgi-beta).
2. Jeśli dysponujemy strukturą przestrzenną białka możemy zintegrować dane sekwencyjne i strukturalne w celu przewidywania epitopów (determinanta antygenowa – fragment antygenu, który łączy się bezpośrednio z wolnym przeciwciałem) pod kątem projektowania przeciwciał. Posiadając dane sekwencyjne i informacje o hydrofobowości i hydrofilowości oraz lokalizację reszt aminokwasowych na powierzchni białka z łatwością można wytypować aminokwasy, które mogą potencjalnie być epitopami strukturalnymi dla przeciwciał.
3. Znajomość powierzchni białka i ładunku elektrostatycznego wraz z przewidywaniami lokalizacji regionów nieuporządkowanych dostarcza cennych informacji o potencjalnych oddziaływaniach białko-białko. Zbiór takich informacji może pomóc w przewidywaniu reszt aminokwasowych na powierzchni, które bezpośrednio będą brać udział w oddziaływaniach.
4. Wysokiej jakości dane strukturalne dają możliwość szczegółowej analizy oddziaływań w kieszeni katalitycznej, np. w celu poznania mechanizmu reakcji. Natomiast, komputerowe analizy związane z dokowaniem molekularnym dostarczają informacji na temat potencjalnych oddziaływań białko-ligand/RNA/DNA.
5. Dzięki wirtualnym badaniom przesiewowym jesteśmy w stanie wyselekcjonować grupy związków chemicznych najlepiej „pasujących” do kieszeni katalitycznej enzymu. Ta metoda jest powszechna we wspieranym komputerowo procesie projektowania nowych leków. Dodatkowo, znając kształt i charakterystykę centrum aktywnego enzymu, możemy zaprojektować takie ligandy, które potencjalnie będą najlepiej blokować aktywność białka. Oczywiście jest to spore wyzwanie, ale oszczędza czas i energię włożoną w badania przesiewowe wykonane w laboratorium.
6. Inżynieria enzymatyczna. W tej dziedzinie przyjmuje się główne założenie, iż skoro głównie struktura białka determinuje jego specyficzną funkcję, to projektowanie nowych funkcji może odbywać się poprzez wprowadzanie ściśle zdefiniowanych zmian w tejże strukturze. I takie zmiany wprowadza się obserwując jaki będą nieść końcowy efekt.
Autor artykułu:
Dr Anna Czerwoniec
VitaInSilica Sp. z o. o.
Krzemowa 1, Złotniki
62-002 Suchy Las
NIP: 9721237412
REGON: 301973876
www.vitainsilica.pl
office@vitainsilica.pl