PRZEDRUK, oryginał dostępny pod adresem www
Fragment skryptu: Spektroskopia IR
Autorzy skryptu: B. Drożdż, M. Tarsa, M. Żylewski
Uniwersytet Jagielloński (www)
Wydział Farmaceutyczny Collegium Medicum (www)
Katedra Chemii Organicznej (www)
Kierownik: Prof. UJ, dr hab. Marek Cegła
Adres:
ul. Medyczna 9
30-688 Kraków
Kontakt: tel. 012 620 55 00
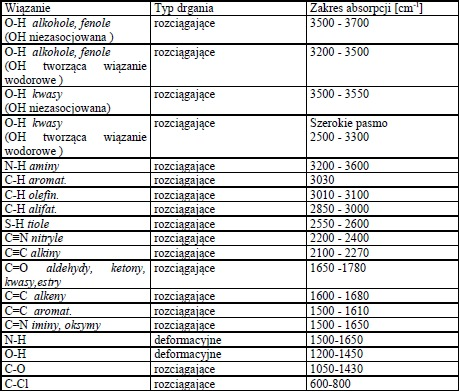
Pełna interpretacja widm IR jest trudna (wręcz niemożliwa), ponieważ w obrębie jednej cząsteczki występuje wiele drgań deformacyjnych i rozciągających. Widmo więc zawiera wiele różnych pasm odpowiadających tym drganiom. Poszczególne rodzaje wiązań mając podobną różnicę energii pomiędzy poziomami oscylacyjnym, absorbują promieniowanie o charakterystycznej częstotliwości, dając pasmo w tym samym zakresie niezależnie od innych szczegółów struktury cząsteczki. Oznacza to, że większość grup funkcyjnych (np. C=O, N-H, O-H) daje charakterystyczne pasma absorpcyjne, których położenie w widmie jest porównywalne. Tabela 1 przedstawia przykłady położenia pasm absorpcji w podczerwieni dla różnych typów drgań i wiązań występujących w związkach organicznych.
Tabela 1. Położenie pasm absorpcji w podczerwieni.
W celu zapamiętania charakterystycznych sygnałów absorpcyjnych w widmie IR można cały zakres podzielić na cztery części.
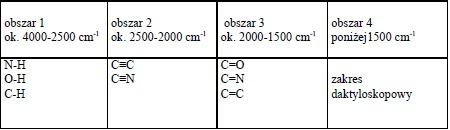
• Obszar 4000-2500 cm-1 odpowiada absorpcji wynikającej najczęściej z obecności w cząsteczce grup N-H, C-H, O-H. Pasma w tym zakresie odpowiadają drganiom rozciągającym.
• Obszar 2500-2000 cm-1 , sygnały w tym zakresie wskazują na obecność w związku grup zawierających wiązania potrójne np. alkiny C≡C, nitryle C≡N . • Obszar 2000-1500 cm-1, pasma w tym zakresie pochodzą głównie od różnego rodzaju wiązań podwójnych (C=O, C=C, C=N).
• zakres poniżej 1500 cm-1 nazwany „zakresem daktyloskopowym” (fingerprint region), posiada układ pasm charakterystycznych dla danej cząsteczki. Są tutaj pasma drgań rozciągających wiązań pojedynczych np. C-C, C-O, C-N oraz wiele pasm odpowiadających drganiom deformacyjnym. Zakres ten wykorzystywany jest do identyfikacji badanej substancji na podstawie porównania jej widma IR z widmem związku wzorcowego, i tak jak w daktyloskopii identyczność zakresu „odcisku palca” stanowi potwierdzenie identyczności badanego związku z wzorcem.
Charakterystyka głównych pasm absorpcji IR wybranych grup związków organicznych
Węglowodory alifatyczne
Alkany
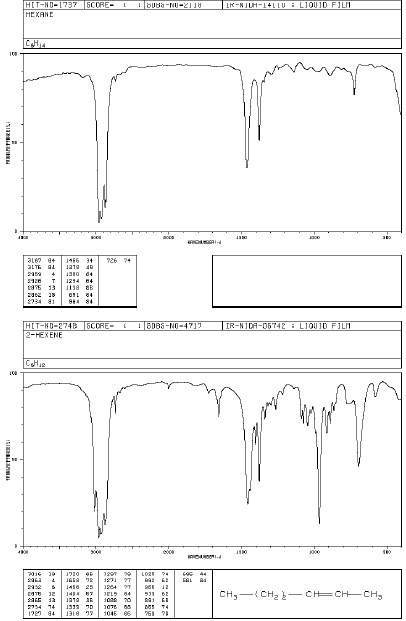
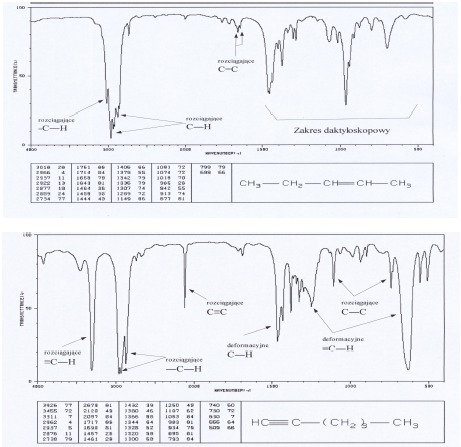
Alkany i cykloalkany wykazują pasma absorpcji w zakresie 2850-3000 cm-1 – odpowiadają one drganiom rozciągającym wiązań C-H i są one najbardziej charakterystyczne i najbardziej przydatne w potwierdzeniu struktury alkanów. Badania dużej liczby węglowodorów nasyconych wykazały obecność dwóch pasm. Pierwsze z nich (ok. 2962 cm-1 pochodzi od asymetrycznych drgań rozciągających, drugie z pasm (ok. 2872cm -1) pochodzi od symetrycznych drgań rozciągających grupy metylowej. Asymetryczne drgania rozciągające i symetryczne drgania rozciągające w grupie metylenowej występują odpowiednio przy 2926 i 2853 cm-1. W widmach węglowodorów alifatycznych i cyklicznych położenie tych pasm nie zmienia się o więcej niż ±10 cm-1. Drgania deformacyjne C-H dają pasma przy 1100- 1300 cm-1. Pasma odpowiadające drganiom rozciągającym C-C są słabe i występują w zakresie 1200-800 cm-1. Drgania te nie mają wielkiego znaczenia dla identyfikacji związku.
Alkeny
W cząsteczce alkenów występują dodatkowo (w porównaniu z alkanami) pasma drgań rozciągających C=C w obszarze 1580-1690 cm-1 oraz drgania rozciągające ( 3010-3095 cm-1) i deformacyjne ( 1290-1420 cm-1) =C-H.
Alkiny
Charakterystyczne pasma absorpcyjne odpowiadające drganiom rozciągającym wiązań C≡C występują w obszarze 2100-2260 cm-1. Pasmo drgań rozciągających ≡C-H leży przy ok. 3300 cm-1 (jest silne i ostre). Drgania deformacyjne ≡C-H dają pasma przy 1220-1370 cm-1 oraz 610-700 cm-1.
Węglowodory aromatyczne
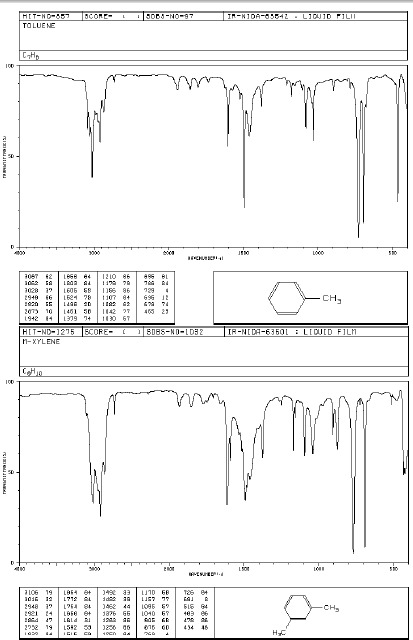
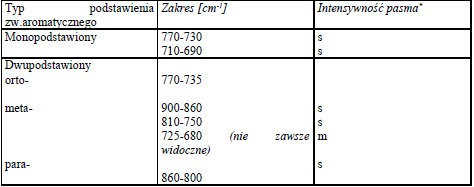
Pasma drgań rozciągających C-H leżą nieco powyżej 3000 cm-1 (mogą być zakryte przez pasma innych ugrupowań występujących w tym zakresie). Serią pasm umożliwiających rozpoznanie struktury aromatycznej są pasma drgań szkieletowych powodujące rozciąganie wiązań C=C w zakresie 1450-1610 cm-1. Ich liczba w zależności od podstawienia dochodzi do czterech. Pasma drgań deformacyjnych C-H występują poniżej 900 cm-1, a ich położenie pozwala ocenić sposób podstawienia pierścienia aromatycznego.
Tabela 2 Drgania deformacyjne wiązań C-H w pochodnych benzenu
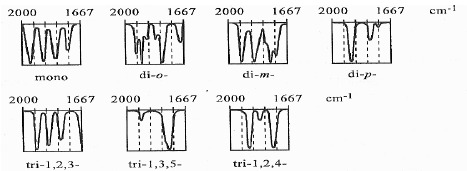
Do oceny typy podstawienia służą również pasma kombinacyjne tonów i nadtonów z zakresu 1600-2000 cm-1 .
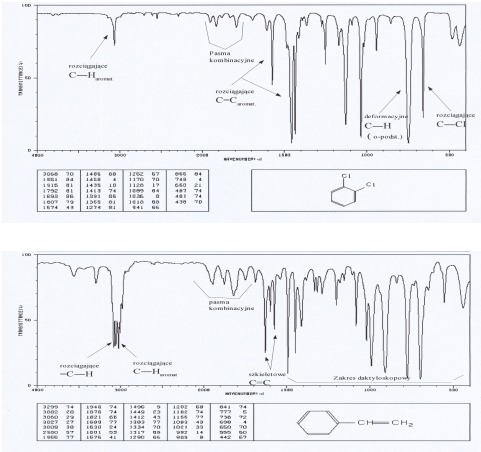
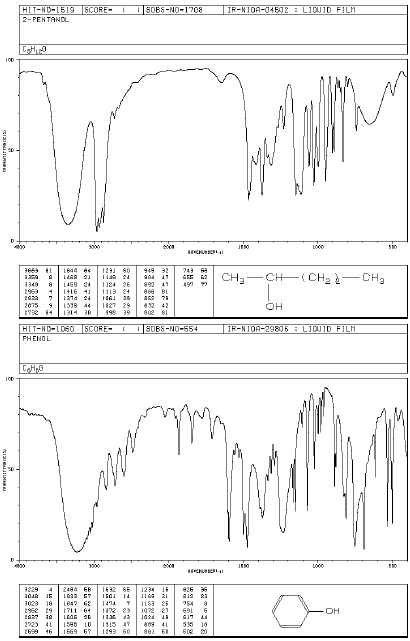
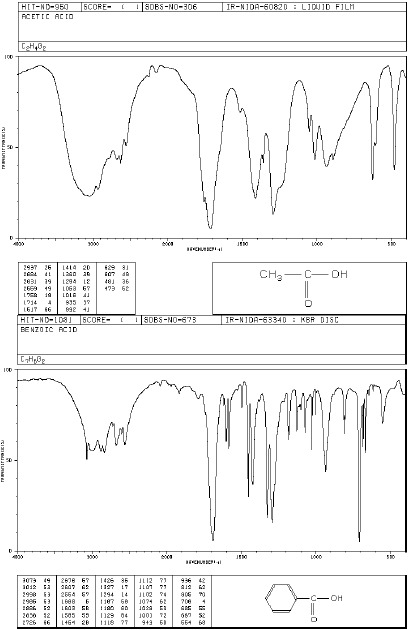
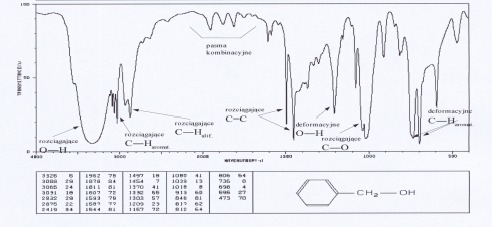
Grupa OH (alkohole, fenole, kwasy)
Grupa O-H w alkoholach i fenolach wykazuje charakterystyczną absorpcję w zakresie 3000- 3600 cm-1. Występują tutaj pasma drgań rozciągających O-H. Swobodnej, niezasocjowanej grupie O-H alkoholi i fenoli odpowiada wąskie pasmo absorpcyjne w zakresie 3580-3670 cm-1. Pasmo to obserwuje się w widmach rozcieńczonych roztworów tych związków. Jednak atom wodoru grupy O-H może łatwo tworzyć wiązanie wodorowe (z atomem tlenu lub z atomami innych grup funkcyjnych). Tworzenie wiązań wodorowych ma wpływ na kształt i położenie pasm absorpcyjnych drgań rozciągających grupy O-H. Tworzenie wiązań wodorowych prowadzi do powstania dimerów i poliasocjatów, wówczas obserwuje się szerokie pasmo w obszarze 3200-3400 cm-1. Kwasy karboksylowe charakteryzują się bardzo szerokim pasmem drgań O-H (związanych z występowaniem wiązań wodorowych), którego maksimum występuje ok.3000 cm-1 a zakres obejmuje 2500-3300 cm-1. Położenie silnych pasm rozciągających C-O zależy od rzędowości alkoholu. Dla I-rzędowych występuje przy 1050-1075 cm-1, dla II-rzędowych przy 1090-1125 cm-1, a dla III-rzędowych przy 1120-1210 cm-1. Fenole badane w stanie stałym (zawiesina, pastylka) absorbują przy 1330-1390 cm-1 oraz 1180-1260 cm-1 . Pasma drgań deformacyjnych O-H leżą w obszarze 1330-1420 cm-1 oraz 650-770 cm-1 i mają małą wartość identyfikacyjną.
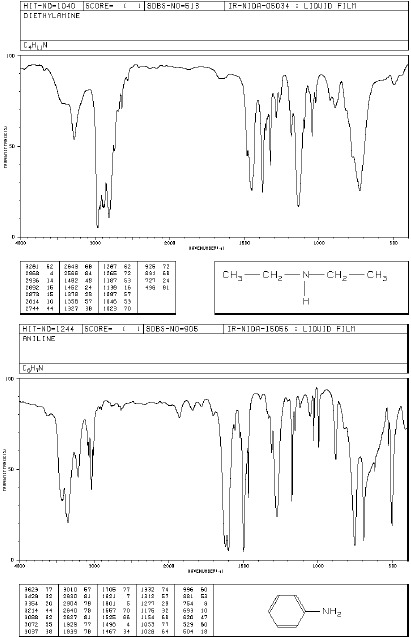
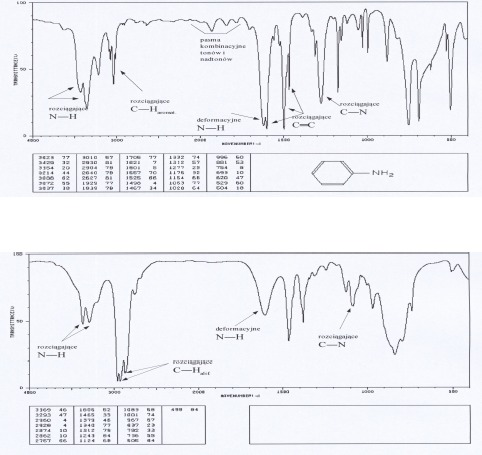
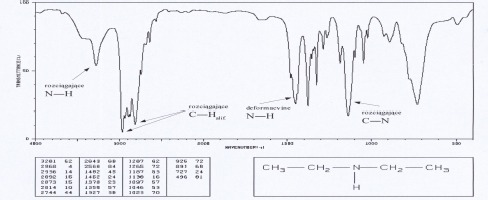
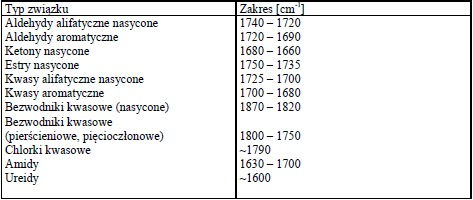
Aminy
Dla amin charakterystyczne pasma absorpcji w podczerwieni występują w obszarze 3300- 3500 cm-1, 1500-1650 cm-1 oraz 1000-1360 cm-1, są związane z drganiami wiązań N-H i C-H. Pierwszorzędowe aminy wykazują dwa pasma w zakresie 3300-3500 cm-1 (drgania rozciągające asymetryczne i symetryczne N-H), aminy drugorzędowe mają tylko jedno pasmo w tym zakresie a dla amin trzeciorzędowych absorpcja w tym zakresie nie występuje. Pasma te dla amin aromatycznych są znacznie intensywniejsze niż dla amin alifatycznych. Obszar 1500-1650cm-1 jest charakterystyczny dla drgań deformacyjnych N-H. Dla amin pierwszorzędowych pasmo drgań deformacyjnych N-H ma duże natężenie (bywa mylone z pasmem drgań rozciągających C=O), dla amin drugorzędowych ma ono mniejsze natężenie. Ciekłe próbki pierwszo- i drgorzędowych amin wykazują średnie lub silne, szerokie pasmo absorpcji w zakresie 660-910 cm-1 , pochodzące od drgań wachlarzowych N-H. Drgania rozciągające C-N dają pasma występujące powyżej 1000 cm-1 . Dla amin alifatycznych pasma te pojawiają się w zakresie 1020-1250, a dla amin aromatycznych 1260-1340 cm-1.
Związki zawierające grupę C=O
W widmach związków karbonylowych pasmo drgań rozciągających C=O występuje w zakresie 1540-1870 cm-1, jest bardzo charakterystyczne ze względu na dużą intensywność. Położenie tego pasma zależy od obecności i usytuowania innych grup funkcyjnych w związku oraz rozpuszczalnika. Jednak dla poszczególnych typów związków pasmo drgań rozciągających C=O jest umiejscowione w dość wąskim zakresie.
Tabela 3 Charakterystyczne zakresy absorpcji w podczerwieni związków z grupą C=O
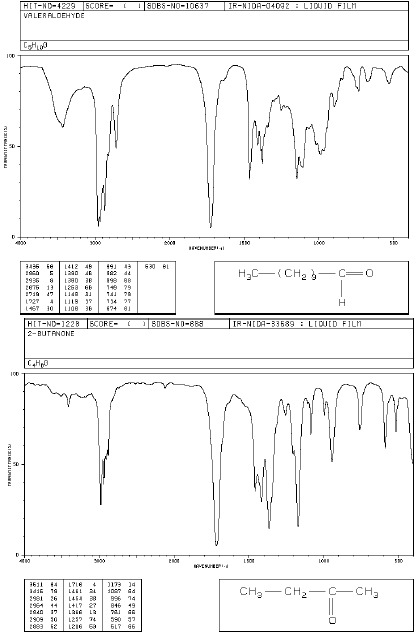
Dla aldehydów oprócz pasm grupy karbonylowej charakterystyczne są pasma związane z absorpcją wiązania C-H grupy funkcyjnej CHO w zakresie 2700- 2830 cm-1, najczęściej w postaci dwóch ostrych pasm 2720 i 2820cm-1 (pasmo to odróżnia aldehydy i ketony).
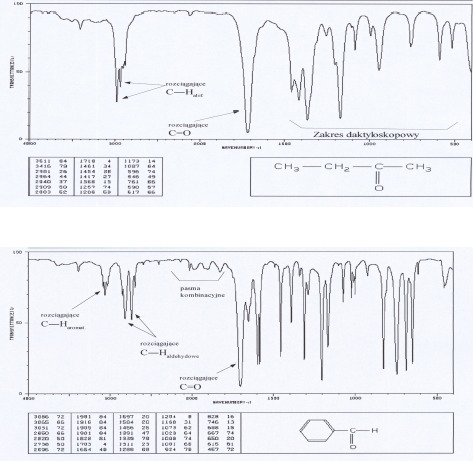

Widma różnych klas związków: