
PRZEDRUK, oryginał dostępny pod adresem www
Spektroskopia magnetycznego rezonansu jądrowego
Autor skryptu: dr Marek Żylewski
Uniwersytet Jagielloński (www)
Wydział Farmaceutyczny Collegium Medicum (www)
Katedra Chemii Organicznej (www)
Kierownik: Prof. UJ, dr hab. Marek Cegła
Adres:
ul. Medyczna 9
30-688 Kraków
Kontakt: tel. 012 620 55 00
Fizyczne podstawy spektroskopii NMR
W spektroskopii magnetycznego rezonansu jądrowego używane jest promieniowanie elektromagnetyczne o częstościach z zakresu radiowego (60 – 900 MHz, choć ciągłe prace nad udoskonalaniem spektrometrów prowadzą do wprowadzania aparatów mierzących przy coraz to wyższych częstościach), które, w porównaniu do stosowanego w innych metodach spektroskopowych, niesie niewielką porcję energii. Może ono zatem wzbudzać przejścia pomiędzy poziomami o niskiej barierze energetycznej, co jest charakterystyczne dla stanów kwantowych związanych z magnetycznymi własnościami jąder atomów budujących cząsteczki chemiczne.
Najprostszym jądrem jest jądro atomu wodoru – proton. Cząstka ta (tak jak każda cząstka elementarna) posiada własność zwaną spinem – wielkością niemającą odpowiednika w fizyce klasycznej. Przybliżając z tym związane zachowanie protonu, wyobrażamy go sobie jako obdarzoną ładunkiem kulkę, wirującą z pewnym momentem pędu. Otrzymuje się w ten sposób klasyczny przykład ładunku w ruchu, który generuje wokół siebie pole magnetyczne, stając się mikroskopijnym magnesem.
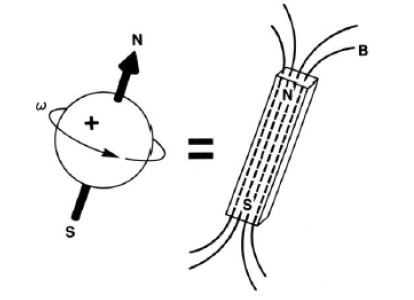
W układzie, w którym nie występuje zewnętrzne pole magnetyczne, orientacja w przestrzeni tak powstałego dipola magnetycznego jest dowolna (brak jest jakichkolwiek oddziaływań). Dopiero obecność zewnętrznego pola magnetycznego powoduje uporządkowanie układu, wymuszając ułożenie dipoli magnetycznych wzdłuż linii tego pola. Analogicznie zachowuje się igła kompasu, będącą małym magnesem sztabkowym, którą można ustawić dowolnie w przypadku braku zewnętrznego pola magnetycznego, natomiast obecność takiego pola wymusza jej ustawienie wzdłuż linii tego pola. Dostarczając energię, można wytrącić igłę kompasu z jej położenia spoczynkowego i obrócić ją o pewien kąt, którego wartość może być dowolna i zależeć będzie od ilości dostarczonej energii i indukcji pól magnetycznych (indukcja pola magnetycznego jest miarą natężenia tego pola) – zewnętrznego (B0) i własnego pola (B) igły kompasu. Podobnie uczynić można z dipolem magnetycznym jądra atomowego (spinem jądrowym), jednakże może on przyjąć tylko ściśle określone orientacje względem linii zewnętrznego pola magnetycznego, związane z odpowiednimi wartościami energii (energia w świecie mikroskopowym jest kwantowana, co oznacza, iż może przyjmować tylko określone wartości). Dozwolone są tylko takie orientacje, dla których rzut spinu (Jz) na kierunek wektora indukcji zewnętrznego pola magnetycznego B0 spełnia warunek:
![]()
gdzie m jest to magnetyczna spinowa liczba kwantowa, mogąca przyjmować wartości od -I do I co 1. I z kolei jest spinową liczbą kwantową, kwantującą wielkość spinu, której wartość jest cechą charakterystyczną dla danej cząstki. Dla protonów (jak i innych cząstek elementarnych budujących atomy) wartość spinowej liczby kwantowej wynosi ½, zatem magnetyczna spinowa liczba kwantowa może przyjąć jedynie dwie wartości: -½ (stan α) i +½ (stan β).
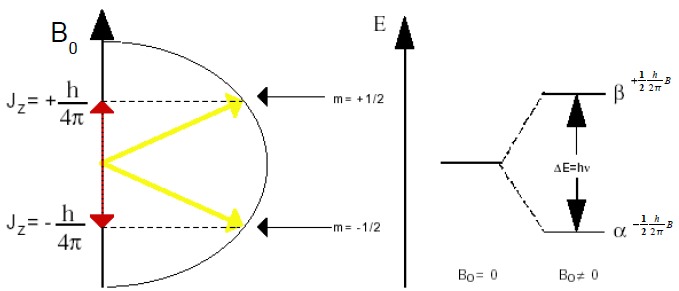
Pojawiają się zatem dwa stany kwantowe, pomiędzy którymi można wzbudzać przejścia energetyczne wskutek absorpcji promieniowania elektromagnetycznego o odpowiedniej energii.
Powyższe rozważania można zastosować również do jąder innych pierwiastków, których jądra atomowe składają się z większej liczby nukleonów. Kwantowa liczba spinowa I jest dla takich jąder cechą charakterystyczną, zależną od ich budowy, jednakże można znaleźć pewne ogólne związki pomiędzy budową jądra atomowego a jego liczbą kwantową.
• Jądra atomowe zbudowane z parzystej liczby protonów i parzystej liczby neutronów mają liczbę I równą 0, zatem magnetyczna spinowa liczba kwantowa przyjmuje też tylko jedną wartość – 0. Konsekwencją jest brak możliwości wzbudzania przejść energetycznych. Wśród jąder ważnych dla analizy związków organicznych są izotopy: węgla 12C i tlenu 16O, dla których nie można otrzymać widm NMR.
• Jądra mające parzystą liczbę nukleonów jednego rodzaju i nieparzystą liczbę nukleonów drugiego rodzaju mają spinową liczbę kwantową równą ½ lub równą wielokrotności liczby ½. Najważniejsze wśród nich są jądra o liczbie I = ½ takie jak 13C (6 protonów i 7 neutronów), 15N, 19F, 31P, które umieszczone w zewnętrznym polu magnetycznym zachowują się analogicznie do protonów i są bardzo cenne z punktu widzenia analizy strukturalnej związków chemicznych.
Spośród innych pierwiastków spotykanych w cząsteczkach związków organicznych obecne są jądra o liczbie kwantowej I = 5/2 jak np. 17O czy I = 3/2 jak np. 35Cl jednakże spektroskopii tych jąder nie stosuje się do prostej analizy strukturalnej ze względu na możliwość wielu przejść energetycznych (dla I = 3/2 występują 4 poziomy energetyczne) bardzo komplikujących obraz widma.
• Jądra zawierające nieparzystą liczbę zarówno protonów jak i neutronów posiadają spinową liczbę kwantową o wartości całkowitej. Najistotniejszym z punktu widzenia spektroskopii NMR jądrem w tej klasie jest jądro deuteru 2D (I = 1), obecne w cząsteczkach rozpuszczalników używanych do wykonywania roztworów próbek, którego sygnał rezonansowy (absorpcyjny) jest sygnałem stabilizacji pola i wzorcowania skali dla współczesnych spektrometrów NMR.
Energia oddziaływania spinu w danym stanie kwantowym z zewnętrznym polem magnetycznym jest dana wyrażeniem:
![]()
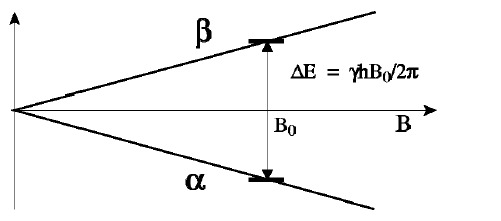
gdzie γ to współczynnik proporcjonalności zwany współczynnikiem magnetogirycznym (funkcjonuje również pojęcie współczynnik giromagnetyczny – charakteryzuje on własności magnetyczne danej cząstki, jądra atomowego). Różnica w energii pomiędzy stanami kwantowymi α i β wynosi:
![]()
Z powyższego równania wynika, iż ilość energii potrzebna do wzbudzenia jądra ze stanu podstawowego, a zatem i częstość promieniowania, jakie może zostać pochłonięte przez próbkę zawierającą dane jądra atomowe, jest proporcjonalna do indukcji (natężenia) zewnętrznego pola magnetycznego i pewnej stałej, charakterystycznej dla danego jądra atomowego.
Umieszczając zbiorowisko jąder atomowych w polu o indukcji 2,3 T, sygnał rezonansowy pojawi się: dla jąder 1H przy 100 MHz, dla jąder 13C przy 25,1 MHz a dla jąder 19F przy 94 MHz. Zmiana indukcji pola magnetycznego pociąga za sobą wprost proporcjonalną zmianę częstości absorbowanego promieniowania i tak przechodząc do pola o indukcji 7,2 T sygnał rezonansowy protonów pojawi się przy 300 MHz a w polu o indukcji 11,7 T przy 500 MHz. Najpotężniejsze współczesne spektrometry NMR pracują przy częstości rezonansowej protonów równej 900 MHz, co jest równoważne polu magnetycznemu o indukcji 20,7 T. Dla porównania można dodać, iż wartość indukcji pola magnetycznego Ziemi na jej powierzchni wynosi ok. 5·10-5 T W próbce badanego związku chemicznego wszystkie jądra danego typu np. protony są identyczne, a zatem zgodnie z powyższym powinny absorbować promieniowanie o tej samej częstości, czyli dać jeden sygnał absorpcyjny, zwany również sygnałem rezonansowym. Należy jednak pamiętać, iż cząsteczki chemiczne to nie tylko zbiorowisko jąder atomowych, ale również ogół elektronów stanowiących zręby atomowe i tworzących różnorakie wiązania chemiczne. Z fizycznego punktu widzenia stanowią one zespół ładunków elektrycznych w ruchu, generujących własne, lokalne pola magnetyczne – Blok. Pola te podlegają sumowaniu wektorowemu z zewnętrznym polem magnetycznym, wpływając na jego natężenie w różny sposób, w zależności od miejsca w cząsteczce – inne pola lokalne będą bowiem generowane przez elektrony zlokalizowane w zrębach atomowych, inne przez elektrony tworzące wiązania typu σ a jeszcze inne przez elektrony tworzące wiązania typu π. Zatem jądra znajdujące się w różnych miejscach cząsteczki, a więc w różnym otoczeniu chemicznym, będą się znajdować pod wpływem różnych wypadkowych pól magnetycznych, co zmieni wielkość bariery energetycznej pomiędzy poziomami i tym samym wpłynie na częstość absorbowanego promieniowania elektromagnetycznego. Obserwując zatem widmo NMR, otrzyma się zestaw sygnałów rezonansowych odpowiadających atomom znajdującym się w różnych miejscach cząsteczki. Te niewielkie różnice w położeniu obserwowanych na widmie sygnałów absorpcyjnych, określa się mianem przesunięcia chemicznego. Wielkość przesunięcia chemicznego zależna jest w sposób ścisły od otoczenia chemicznego danego jądra, czyli od rodzaju, krotności i ilości wiązań chemicznych i rodzaju i ilości atomów będących w bezpośrednim otoczeniu badanego jądra. Ponieważ zarówno bariera energetyczna pomiędzy poziomami energetycznymi jąder, jak i natężenie lokalnych pól magnetycznych zależy od wielkości B0, położenia sygnałów rezonansowych na widmie nie można przedstawiać podając bezwzględnych wartości częstości (czy też długości fali) absorbowanego promieniowania elektromagnetycznego – taki sposób prezentacji widm dawałby różne rezultaty w zależności od konstrukcji aparatu, a ściśle rzecz biorąc w zależności od indukcji używanego w nim pola magnetycznego, co powodowałoby brak możliwości porównywania wyników eksperymentów wykonanych w różnych laboratoriach. Z tego powodu w spektroskopii NMR stosuje się pomiar względny – wykonując widmo dla danego jądra, wprowadza się substancję wzorcową, dla której sygnał rezonansowy badanych jąder atomowych wyznacza punkt 0 na skali, natomiast położenie sygnałów rezonansowych próbki podawane jest względem sygnału rezonansowego wzorca w następujący sposób:
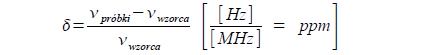
Powstaje w ten sposób skala przesunięć chemicznych δ, która zgodnie z podanym wyżej wzorem jest skalą bezwymiarową. Ze względu na bardzo małą wartość różnicy pomiędzy częstością rozpatrywanego sygnału próbki a częstością sygnału wzorca (pola lokalne powodujące wystąpienie zjawiska przesunięcia chemicznego są bardzo małe w porównaniu z polem B0) wyraża się ją w Hz natomiast częstość wzorca w mianowniku w MHz (106 Hz). Otrzymana w ten sposób skala przesunięć chemicznych odpowiada liczbowo milionowym częściom wartości częstości sygnału rezonansowego obserwowanego jądra. Zaznacza się to w opisie skali skrótem ppm (part per million). Tradycyjnie skalę przesunięć chemicznych obliczyć również można podając wartości zarówno w liczniku, jak i mianowniku powyższego ułamka w Hz i mnożąc wynik przez 106. Obserwując położenie sygnałów rezonansowych na widmie, otrzymuje się informację na temat otoczenia chemicznego jąder atomowych w cząsteczce, co pozwala na określenie jej struktury chemicznej.
Charakterystycznym przykładem zależności pomiędzy otoczeniem chemicznym a obserwowanym sygnałem rezonansowym jest wpływ obecności układu aromatycznego na położenie na widmie sygnału rezonansowego protonów. Sekstet elektronów π zachowuje się w zewnętrznym polu magnetycznym B0 jak obracająca się pętla przewodnika. Zgodnie z zasadami elektrodynamiki w takim przewodniku generowany jest przepływ prądu (powstanie tego prądu zwanego prądem pierścieniowym jest obecnie uważane za jedno z najlepszych kryteriów świadczących o aromatyczności związku), który z kolei powoduje powstawanie lokalnego pola magnetycznego Blok, skierowanego przeciwnie do pola zewnętrznego wewnątrz pierścienia a zgodnie z nim na zewnątrz pierścienia.
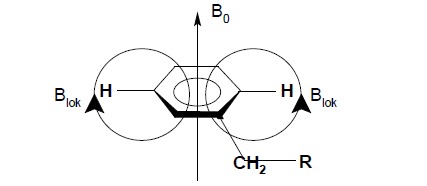
Protony związane z pierścieniem aromatycznym będą podlegały działaniu pola B=B0+Blok, podczas gdy protony nie związane bezpośrednio z pierścieniem np. protony łańcucha węglowego takiego wpływu pola lokalnego pochodzącego od układu elektronów π odczuwać nie będą. Sygnał rezonansowy protonów aromatycznych pojawi się zatem przy ok. 7,3 ppm natomiast sygnał protonów alifatycznych obserwowany będzie przy wartościach mniejszych niż 2,5 ppm.
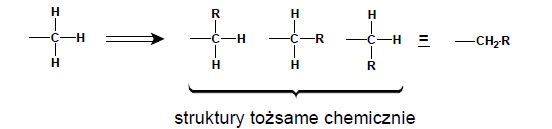
Ze względu na występowanie zjawiska przesunięcia chemicznego dla atomów posiadających różne otoczenie chemiczne na widmie NMR pojawiać się będą różne sygnały rezonansowe. Ilość tych sygnałów jest kolejnym bardzo ważnym parametrem pomagającym w określeniu struktury chemicznej badanego związku. Aby poprawnie przewidzieć ilość sygnałów obserwowanych na widmie i odwrotnie, aby z ilości sygnałów móc określić ilość grup różnych atomów w cząsteczce, należy rozpatrzyć problem równocenności atomów. Najprostszym kryterium określania równocenności atomów jest tzw. kryterium podstawienia. Rozpatrując atomy wodoru w grupie metylowej stwierdzić można, iż są one nierozróżnialne pod względem chemicznym. Kryterium podstawienia stwierdza, iż podstawienie któregokolwiek z nich da zawsze tą samą pochodną:
Podobnie uznać można, iż wszystkie atomy wodoru np. w benzenie są tożsame chemicznie, ponieważ podstawienie któregokolwiek z nich np. atomem chloru da w rezultacie zawsze chlorobenzen, niezależnie od tego który z atomów wodoru zostanie podstawiony. Zatem atomy wodoru w grupie metylowej są równocenne i podobnie wszystkie atomy wodoru w benzenie są równocenne. Jednocześnie równocenne atomy mają zawsze takie samo otoczenie chemiczne, więc dają zawsze jeden sygnał na widmie. Kryterium podstawienia zastosować oczywiście można także dla bardziej skomplikowanych przypadków. Dla zobrazowania przedstawiono poniżej wyznaczenie grup równocennych atomów wodoru dla metoksybenzenu:
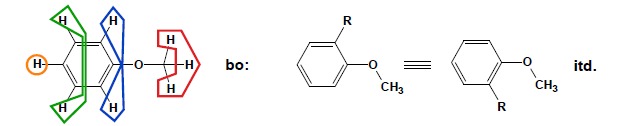
Obserwuje się tutaj 4 grupy równocennych atomów wodoru (w każdej z tych grup spełnione jest kryterium podstawienia – np. podstawienie któregokolwiek z protonów w pozycji orto oznaczonych kolorem niebieskim da taką samą pochodną więc są one równocenne) zatem na widmie 1H NMR spodziewać się można obecności 4 sygnałów rezonansowych. Rozumowanie to oczywiście można przenieść również na atomy innych pierwiastków.
Praktyczne aspekty wykonywania widm NMR
Uproszczony schemat ideowy współczesnego spektrometru NMR przedstawiono poniżej:
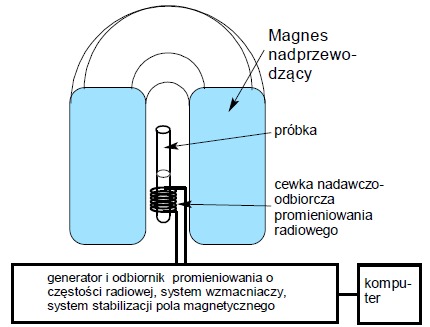
Pole magnetyczne generowane jest przez elektromagnes nadprzewodzący, którego cewka chłodzona jest ciekłym helem (temp. -270°C). Umożliwia to uzyskanie pól magnetycznych o bardzo wysokiej indukcji. Próbka umieszczana jest w szklanej kuwecie, wewnątrz magnesu, w sondzie pomiarowej, której najważniejszym elementem jest cewka nadawczo-odbiorcza promieniowania radiowego. Obecnie spektrometry NMR pracują techniką impulsową, w której na próbkę działa krótki impuls promieniowania elektromagnetycznego tak dobrany, aby wzbudzeniu uległy wszystkie obserwowane jądra atomowe (np. wszystkie protony w próbce, niezależnie od ich otoczenia chemicznego). Następnie cewka pomiarowa rejestruje odpowiedź próbki w trakcie jej powrotu do stanu podstawowego. Otrzymany sygnał jest następnie przetwarzany przez komputer na postać widma NMR na drodze tzw. szybkiej transformacji Fouriera.
Widma NMR na ogół rejestruje się w roztworach (choć możliwa jest również rejestracja widm NMR w ciele stałym przy użyciu specjalnych technik pomiarowych). Rozpuszczalnik należy tak dobrać, aby nie zakłócał obrazu widma. Ponieważ najczęściej wykonuje się widma 1H NMR, w stosowanych rozpuszczalnikach eliminuje się atomy wodoru poprzez ich wymianę na cięższy izotop – deuter. Do najczęściej stosowanych rozpuszczalników należą: CDCl3, DMSO-d6, D2O (ciężka woda). W wyniku pomiaru otrzymuje się widmo, którego istotne dla analizy elementy przedstawiono poniżej:
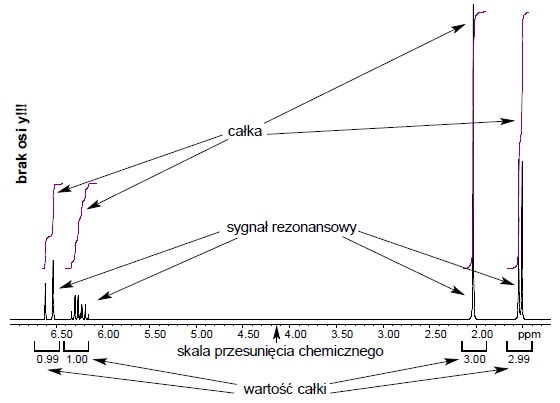
Należy zwrócić uwagę na brak osi Y na widmie. Wysokość sygnałów w spektroskopii NMR nie ma żadnego znaczenia, zatem osi Y na widmach się nie umieszcza. Miarą intensywności sygnałów jest ich pole powierzchni – czyli z matematycznego punktu widzenia – całka.